Growing String Method
Note
gsm is not developed in our group but in the ZimmermanGroup, therefore this tutorial and the useage of GSM is without warranty of completness or correctness.
gsm is not able to communicate with xtb, therefore a fake orca output is created using the xtb values.
To run a gsm calculation, the following programs / files are needed.
gsm.orca in any valid path, e.g. your bin
inpfileq in the directory, where you want to execute your calculation
modified ograd in the directory, where you want to execute your calculation
tm2orca.py in any valid path, e.g. your bin
Those are distributed from our fork of gsm here for your convenience.
For further information and a detailed description on gsm, see ZimmermanGroup and their orca interface.
Input
gsm is a method to find a reaction path and a transition state. In the following, we are going to work with the Double-Ended Growing String Method (DE-GSM), therefore a converged start and end structure is needed. The atomic order needs to be the same in both files, otherwise the gsm calculation will not give the transition state you are looking for.
In your current working directory cwd/ you need to have the files inpfileq and ograd, and the directory scratch/. In scratch/, all files created and needed during the calculation are saved, but in the beginning, there is only one file in scratch/, named initial0000.xyz. The file initial0000.xyz contains the start and the end structure in any valid Xmol format. With the inpfileq the user is able to set specific parameters for the gsm calculation, whereas the ograd wraps xtb and converts input and output for gsm. This is necessary, as gsm can read orca output but not xtb output. To use gsm with xtb, we therefore have to fake an orca output, which is done using the tm2orca.py script.
> cd cwd/
> ls
inpfileq ograd* scratch/
Inversion
This example deals with the inversion of cyclohexane from the chair to the boat conformation. Firstly, you need a converged starting structure start.xyz. You therefore have to build your molecule using a smiles string, avogadro or any other graphical program of your choice. Afterwards you have to execute a quick geometry optimization (e.g. with xtb) and copy the obtained converged structure back in your cwd/.
> xtb unconverged.xyz --opt
> cp xtbopt.xyz ../start.xyz
Obviously, the xtb calculation can be done using all flags explained in this documentation, e.g. –chrg, –uhf, –alpb and so on.
The same has to be done with the end structure. It is advisable to take the optimized start structure and change, whatever you want to change, using a graphical program, which does not change your atomic order, e.g. avogadro, as a change in the atomic order will definitively cause problems during the DE-GSM calculation!
Before you can start the calculation, a couple of other things have to be done. First, you have to generate your initial0000.xyz.
> cat start.xyz end.xyz > scratch/initial0000.xyz
> cat scratch/initial0000.xyz
18
C 0.72407050811334 1.25235108200516 -0.24338094705465
C -0.72343762657313 1.25319617766380 0.23929059852918
C -1.44731174776017 -0.00012207898744 -0.24387881064420
C -0.72448059275055 -1.25351379257321 0.24067027167326
C 0.72302665800789 -1.25441395902634 -0.24200633751896
C 1.44695184726671 -0.00106936630608 0.24102971332150
H 0.74079379322633 1.27977999101331 -1.33568337236737
H 1.23911262989111 2.14407418287692 0.12103331256485
H -1.23773720847930 2.14497568327721 -0.12601425207736
H -0.74014360366626 1.28171032471934 1.33155904823478
H -2.47725186596728 0.00050340452584 0.12007291068245
H -1.47894487592394 -0.00071222738449 -1.33620018565333
H -0.74120355748858 -1.28080687484113 1.33297520098609
H -1.23953398541620 -2.14527362819323 -0.12363941525185
H 1.23731965319244 -2.14616191699557 0.12338823394548
H 0.73972201150389 -1.28305135703018 -1.33427294070718
H 1.47879051289637 -0.00048052377193 1.33334318895494
H 2.47682744992731 -0.00169512097196 -0.12311621761763
18
C 0.73801367871811 1.26986541848913 -0.29891956390957
C -0.72425034407001 1.23660126909089 0.13751131482082
C -1.44534047314084 -0.03177648038732 -0.35671028842475
C -0.51312926640361 -0.95386524177349 -1.13921033932034
C 0.73874270548611 -1.27320174695686 -0.32527750643084
C 1.46163460859193 0.00218599807452 0.14815613748902
H 0.80167461165837 1.37397900540374 -1.38290508569736
H 1.22823701649042 2.14109015941832 0.14110672721895
H -1.22954714616749 2.12895335124983 -0.23651035666162
H -0.75981821609326 1.27774751863575 1.22804091215143
H -1.84194534594910 -0.58465393222957 0.49789315375467
H -2.29332568018213 0.23757670740058 -0.98938629993452
H -1.03732116020641 -1.88207230826469 -1.37787118249708
H -0.23326413070136 -0.48637114937456 -2.08427188018927
H 0.44512664267991 -1.87161892139158 0.54024210888424
H 1.41101252131720 -1.88609677273023 -0.92920597460851
H 1.52271848431407 0.00555688995561 1.23838178865502
H 2.48432149365809 0.02132023538992 -0.23310366530028
Then you have to modify your inpfileq. Normally, all default values can be used, and you only have to care about the last two entries TS_FINAL_TYPE and NNODES. TS_FINAL_TYPE can be 0 or 1. 0 means no bond breaking and is used for this inversion, whereas you have to use 1 for a bond breaking. If you use the wrong setting here, so in this case 1 for the inversion of cyclohexane, gsm tries to break a bond leading to a wrong path. NNODES is the maximum number of nodes for the DE-GSM calculation and should be set to at least 15 for xtb.
TS_FINAL_TYPE 0 # 0=No bond breaking, 1=breaking of bond
NNODES 15 # including endpoints
Last, you have to modify the xtb call in ograd*. The $ofile.xyz as well as the –grad flag are necessary, but you can modify e.g. your charge or alpb flag. In the case of cyclohexane, the charge is 0 and for simplifications I just calculate it in gasphase, therefore no ALPB is used.
xtb $ofile.xyz --grad --chrg 0 > $ofile.xtbout
Now, you have done everything to start the calculation.
> gsm.orca
After the calculation, the two most important files are the reaction path in your cwd/, called stringfile.xyz0000, and the transition state in scratch/tsq0000.xyz, both in a valid Xmol format. The reaction path of the Inversion of cyclohexane can be seen below.
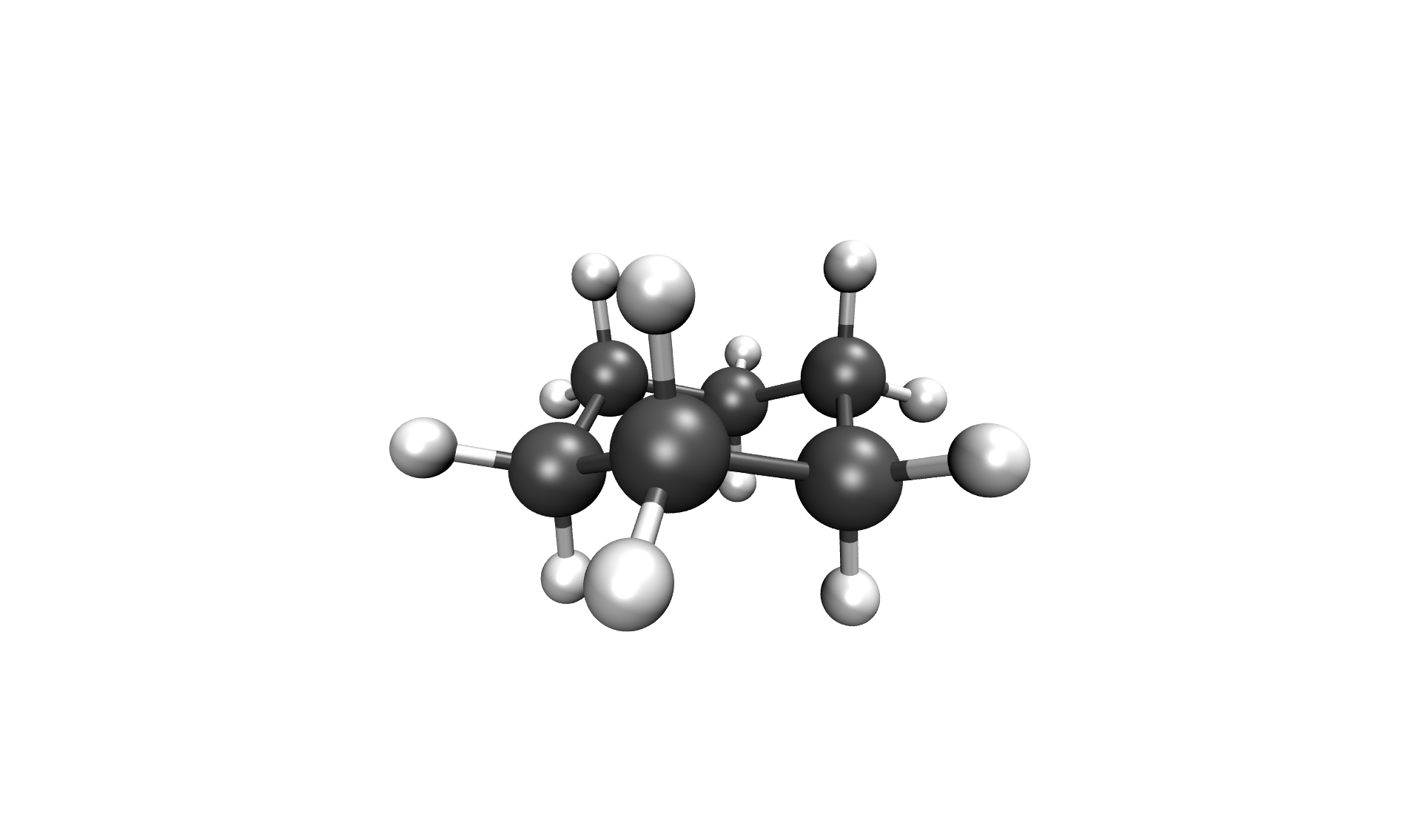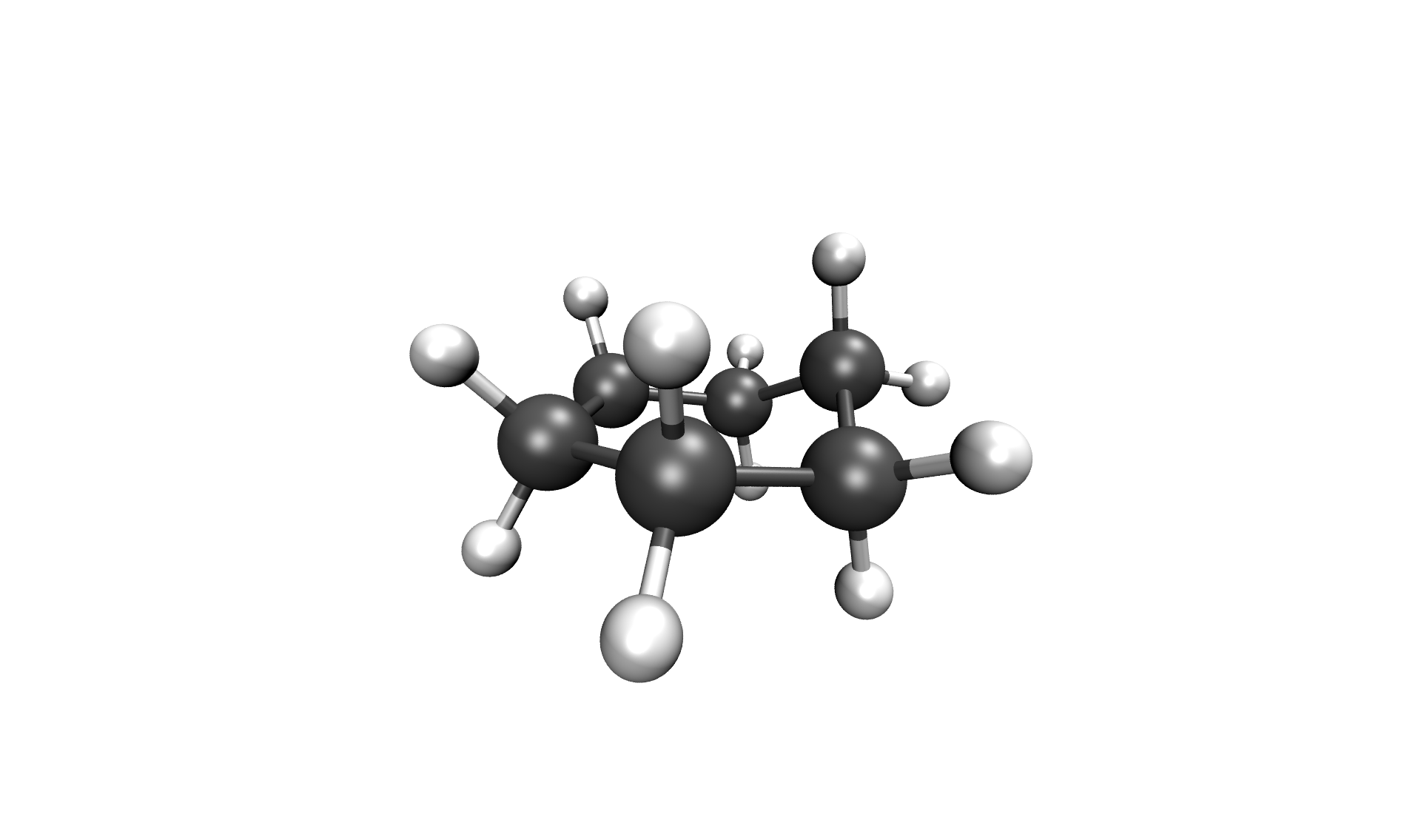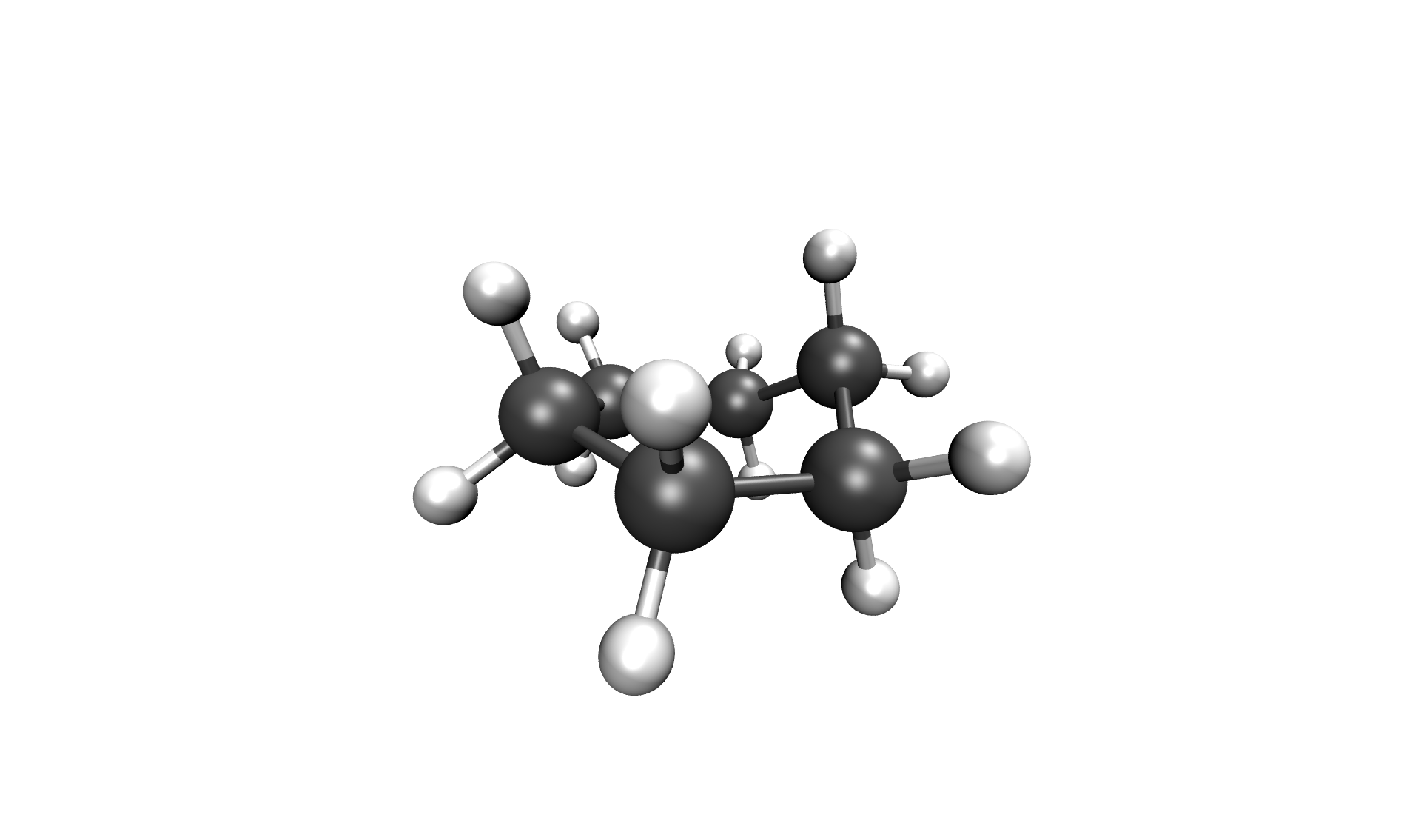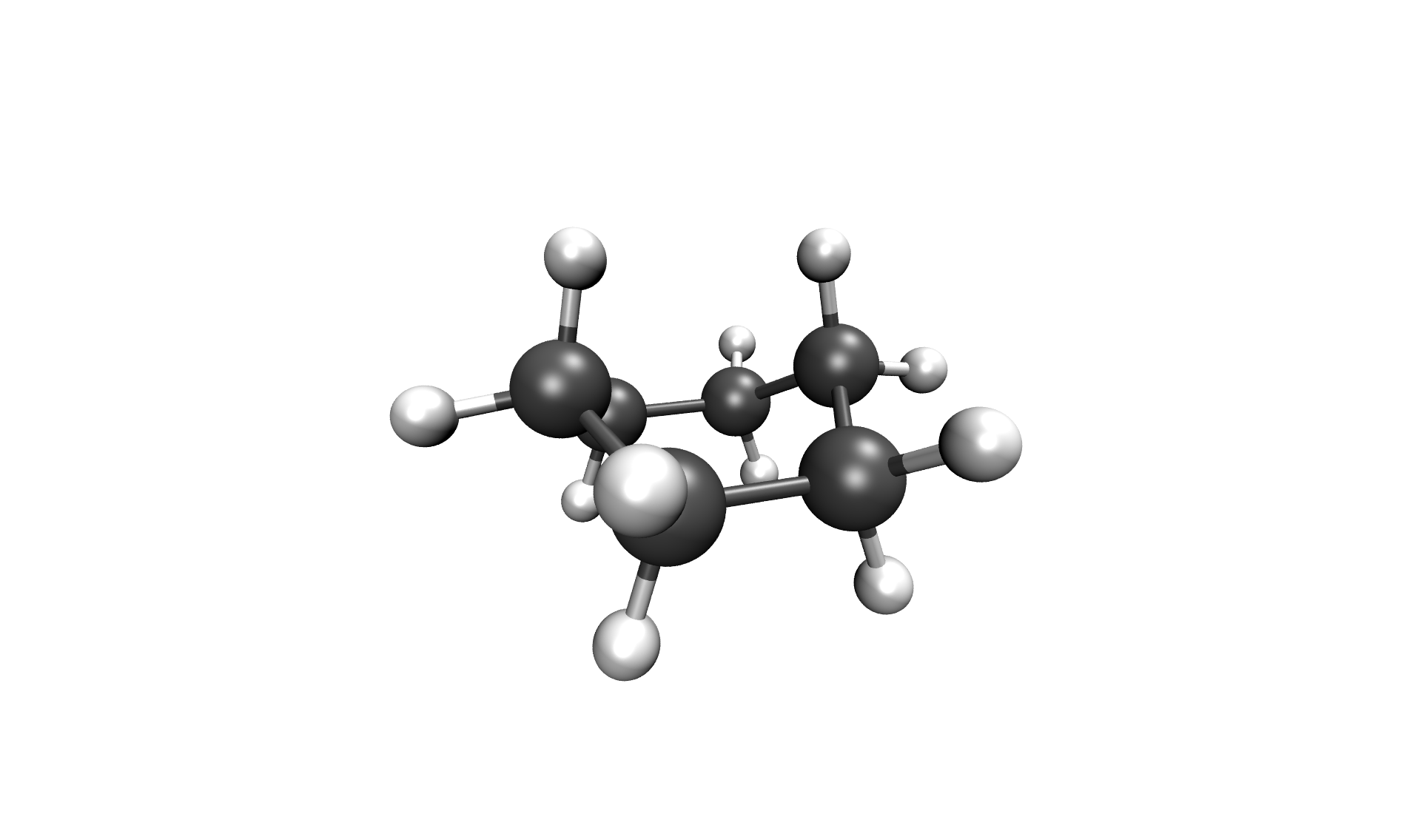
Inversion of cyclohexane

Energy diagram of the inversion of cyclohexane
Bond breaking
The next example is a simple Claisen rearrangement of an allyl vinyl ether and consequently includes a bond breaking and building. The initial0000.xyz is build as described above by writing the converged start and end structure one after the other.
> cat start.xyz end.xyz > scratch/initial0000.xyz
> cat scratch/initial0000.xyz
14
C 0.33830681 -0.40028145 0.06863012
C 0.10595161 -0.26718767 1.36421188
H 1.33077226 -0.61906183 -0.27493881
H -0.42216146 -0.28728678 -0.68244497
O -1.06599246 -0.01419187 2.00107453
H 0.89080386 -0.36692363 2.10223944
C -2.24339525 0.08535540 1.21865884
H -3.06296651 0.00347496 1.94095352
C -2.38810216 1.37002374 0.45318426
H -2.30704191 -0.76808842 0.53050462
H -3.21531691 1.36845744 -0.24273208
C -1.61866094 2.43160218 0.59926563
H -0.79697159 2.43969569 1.29630648
H -1.77723230 3.33005423 0.02997950
14
C 0.05083404 0.47756955 0.03067754
C 0.22099793 -0.53384083 1.12248949
H 1.00063556 0.99546491 -0.11008883
H -0.23550427 -0.01507412 -0.90051555
O -0.06214314 -1.70052772 1.01406801
H 0.61484477 -0.11647527 2.06863484
C -3.09105601 0.69502179 1.56213016
H -4.07672239 0.25168355 1.53446340
C -2.38605593 0.89986170 0.46164886
H -2.72406577 0.97143579 2.54163695
H -2.77578741 0.61350077 -0.51143129
C -1.01585926 1.51412664 0.44531292
H -0.76139644 1.92312285 1.42742393
H -0.99072867 2.32977240 -0.28155745
Next, the inpfileq is modified. As we are now dealing with a bond breaking, the TS_FINAL_TYPE has to be adapted. The NNODES is also changed to a higher value to give a more detailed reaction path. This is not necessary and was just done to play a bit with the settings.
TS_FINAL_TYPE 1 # 0=No bond breaking, 1=breaking of bond
NNODES 20 # including endpoints
At the end, the ograd* has to be modified. As Claisen rearrangements are often done in polar solvents, the calculcation was done using ALPB(water).
xtb $ofile.xyz --grad --chrg 0 --alpb h2o > $ofile.xtbout
tm2orca.py $basename
Now, the gsm calculation is done
> gsm.orca
The reaction path as well as the energy diagram are given below.
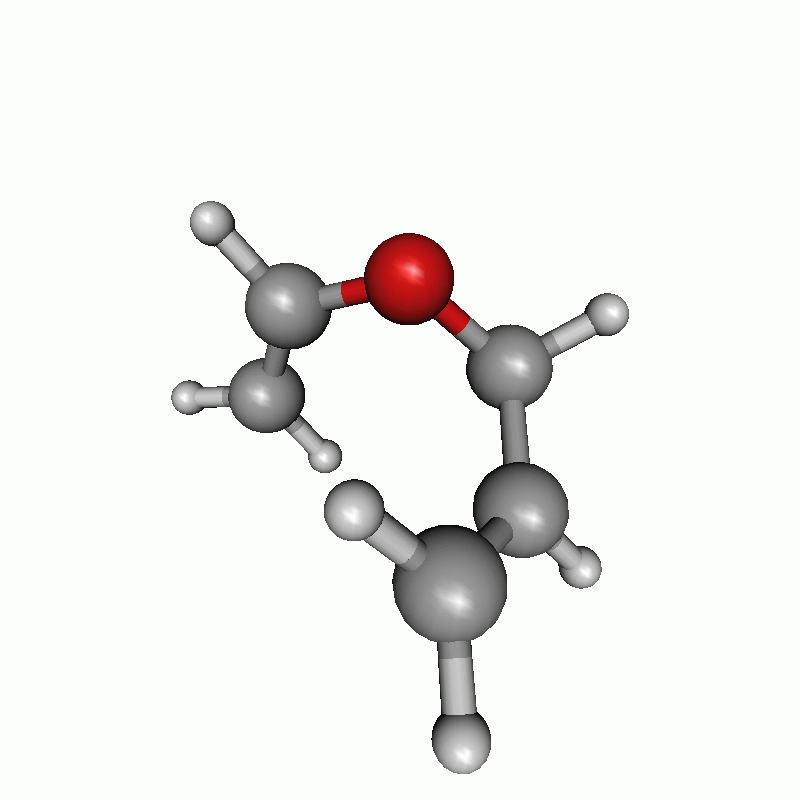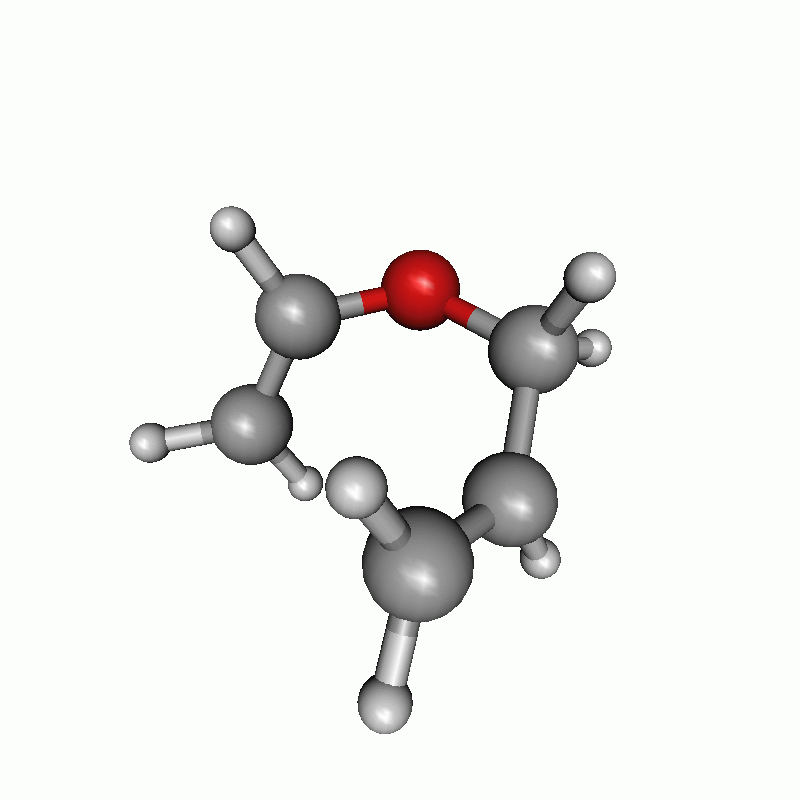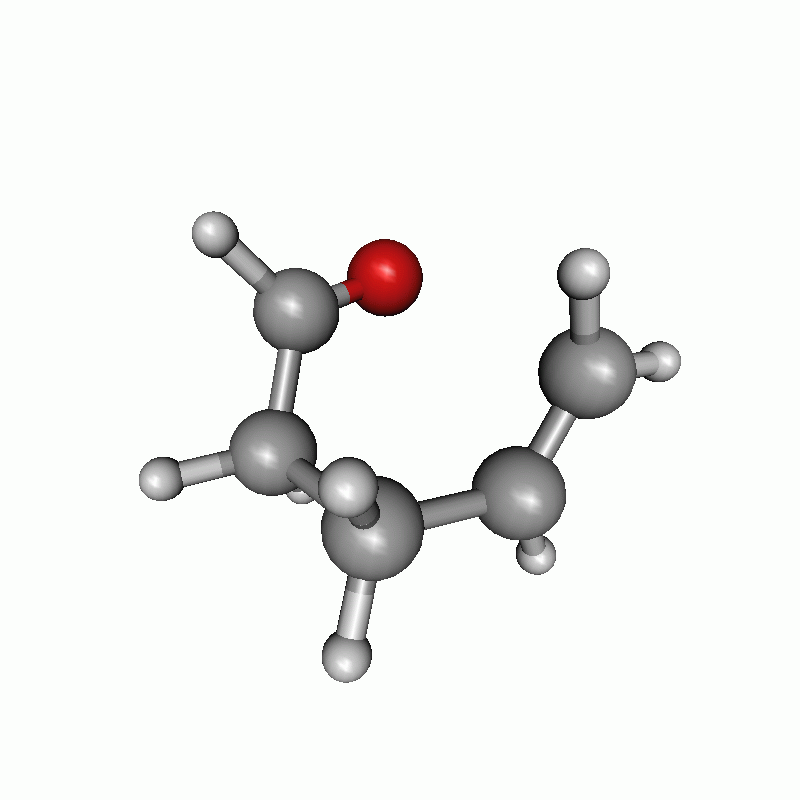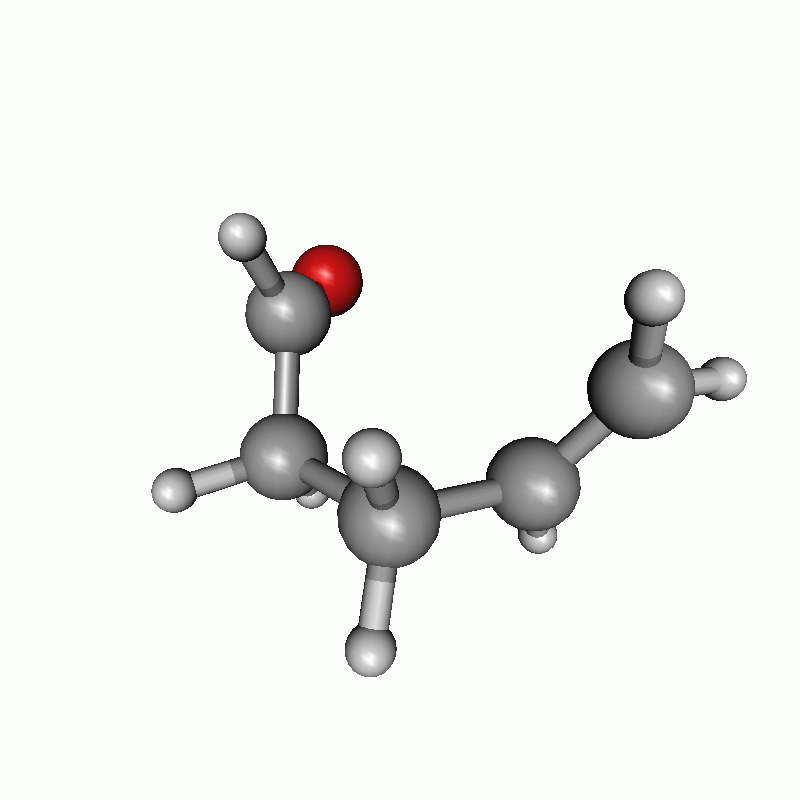
Reaction path of a claisen rearrangement
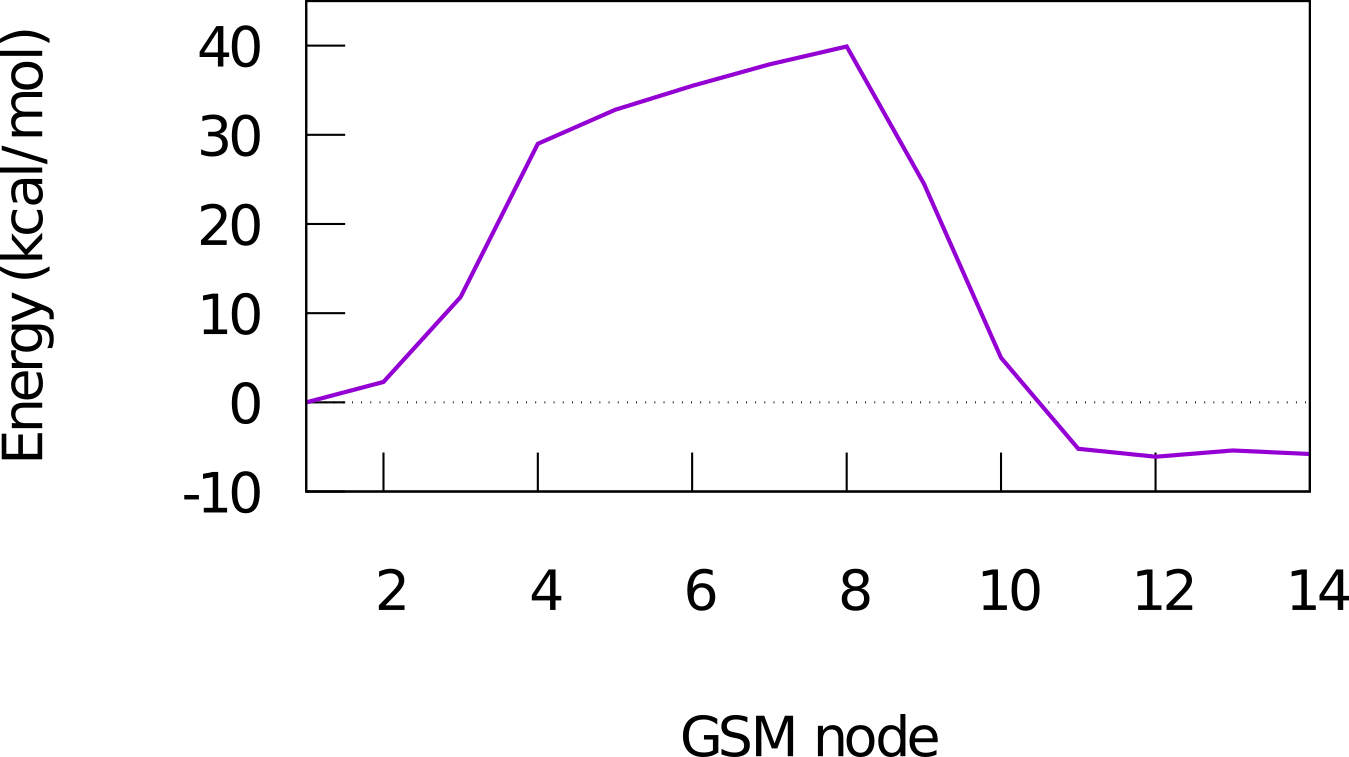
Energy diagram of a claisen reaction
Wrong atomic order
The following is an example that shows how important a proper atom order is. It deals with the same Claisen rearrangement as shown above, but with a different atom order in the start and end structure file, as shown below.
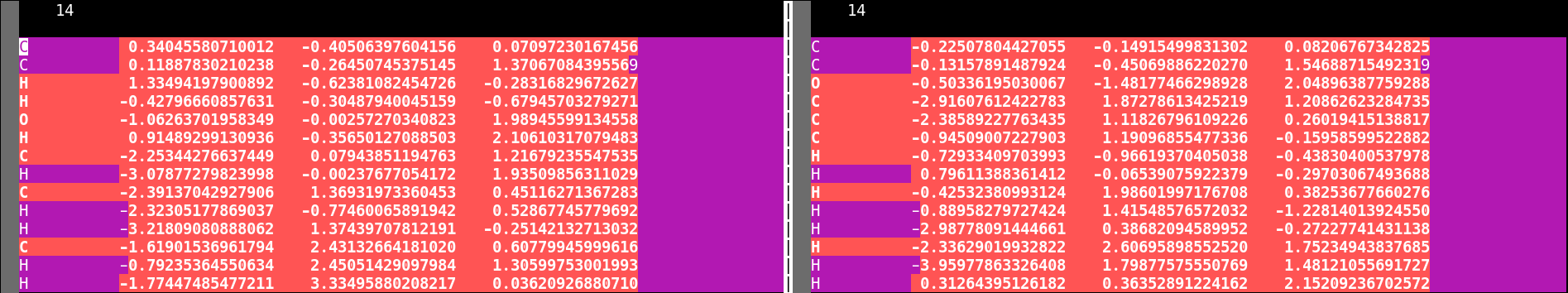
vimdiff of different atomic order in the start (left) and end (right) file
Except for the different atom order the same as above was done. Both structures are written to the initial0000.xyz in the scratch/* directory. In the inpfileq the TS_FINAL_TYPE is 1, and the NNODES is set to 20. The xtb call in ograd* is given below:
xtb $ofile.xyz --grad --chrg 0 --alpb h2o > $ofile.xtbout
Now gsm is just started as already shown.
> gsm.orca
The resulting path as well as the energy diagram is shown below.
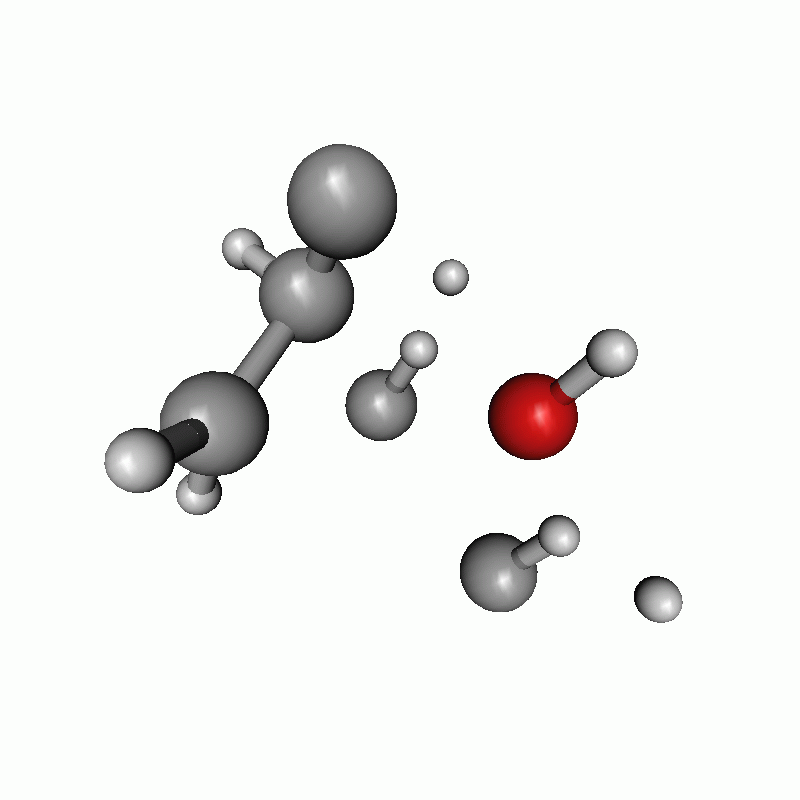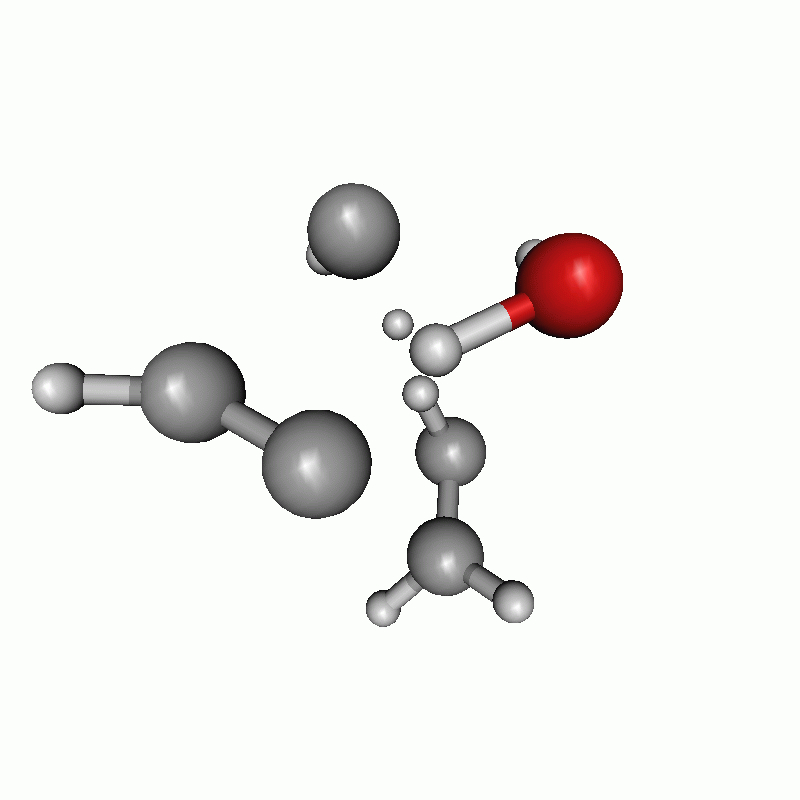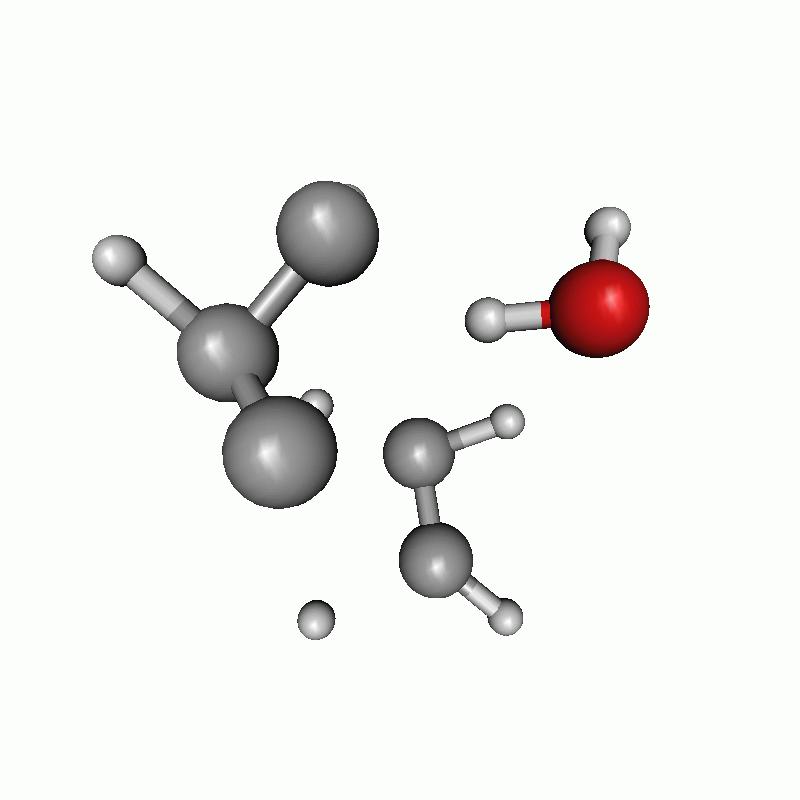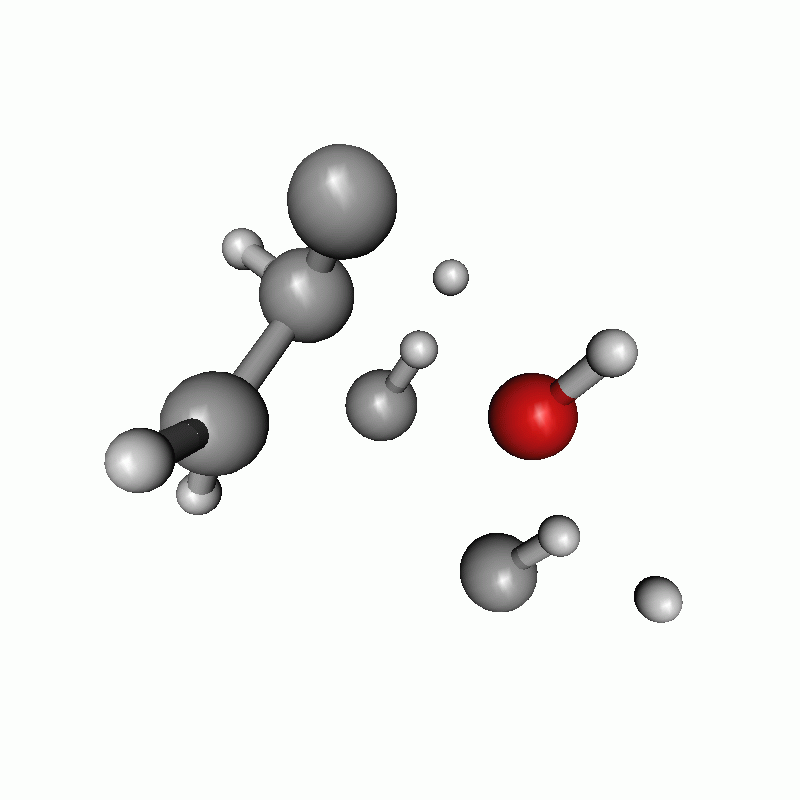
Reaction path of a claisen rearrangement with wrong atom order
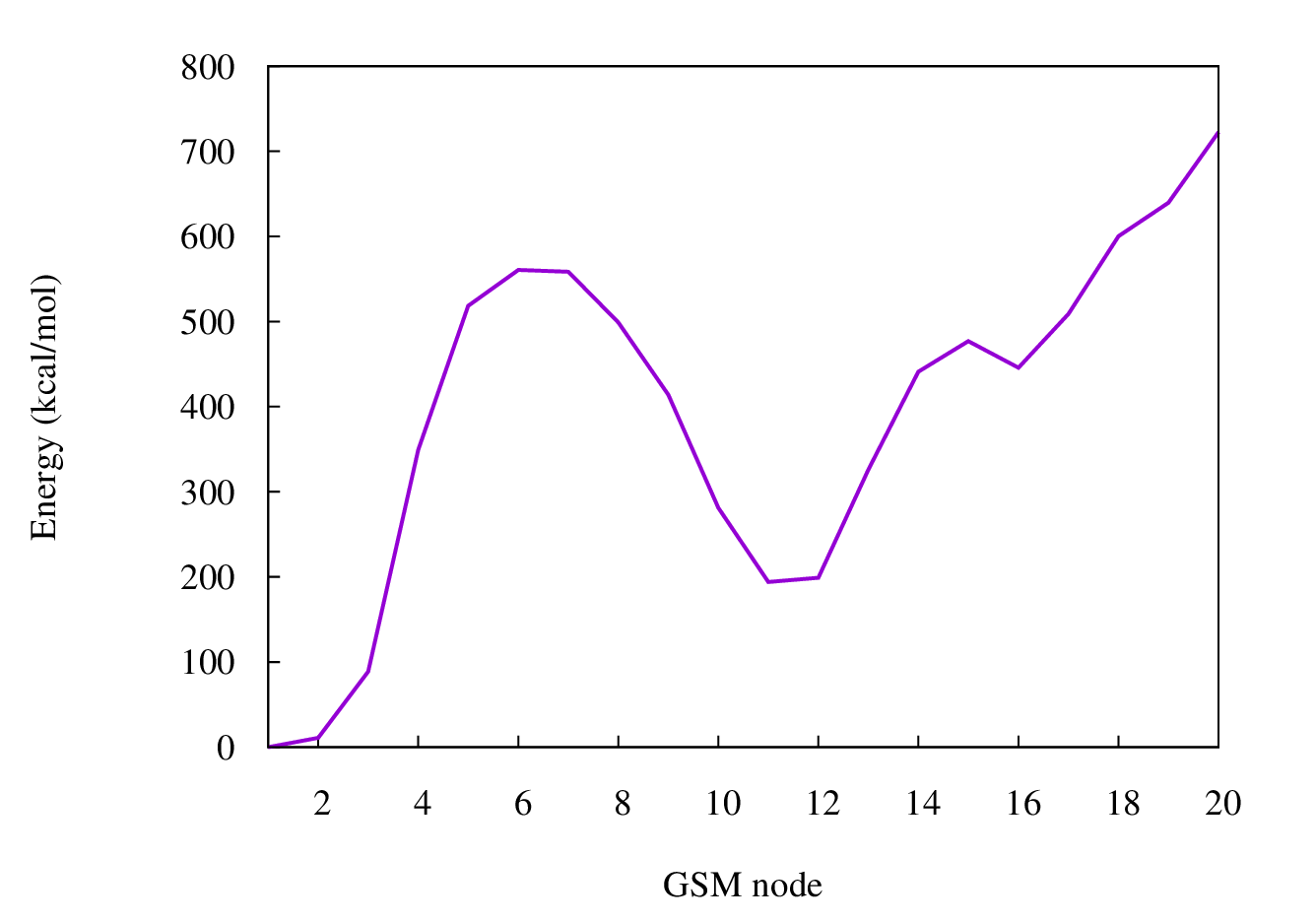
Example of an energy diagram of a wrong reaction path