DIPRO
This guide aimes to give a general overview over the DIPRO implementation within the xtb program package.
Note
This feature is only present in the current bleeding-edge version.
General description
The dimer projection method, known as DIPRO (Andrienko2010), enables the calculation of the electronic coupling integrals between pairs of molecules. DIPRO necessitates three quantum mechanical single-point calculations from any source (e.g., SQM, HF, DFT): one for each monomer (A,B) and another for the dimer (AB), whereas the optimized monomer structures are used in the unrelaxed dimer geometry. The relevant equations are provided below:
where \(C\) are the orbital coefficients, \(S\) is the AO overlap matrix, \(E\) are the orbital energies, \(i\) and \(j\) denote the molecular orbitals that are considered for the electron or hole transfer. Use the coupling integrals on your way to estimate conductivity and charge transport in materials.
Input
To perform a DIPRO calculation with xtb the --dipro option is used. You can specify a threshold (optional) within which energy range around HOMO and LUMO you want orbitals to be included into the DIPRO run. Default is 0.1 eV.
> xtb <geometry_file> --dipro 0.1
Functionality
runtype
The DIPRO runtype offers similar options like the SCC runtype. Feel free to use solvent, electronic temperature, charge, UHF, method … as usual. DIPRO is not restartable from xtbrestart because three singlepoints in a row are performed.
flags
- --chrg ‘int’:
You can use the classical
--chrgflag to provide the charge for the dimer system. DIPRO will assign it automatically to the individual fragments. An alternative is to provide a .CHRGfrag file and specify the charges for both fragments in the formINT INT.- --uhf ‘int’:
You can use the classical
--uhfflag to provide the number of unpaired electrons for the dimer system. As DIPRO cannot assign it automatically to the individual fragments, you must provide a .UHFfrag file and specify the number of unpaired electrons for both fragments in the formINT INT.
Note
If .UHFfrag is not given, the calculation will abort.
xcontrol
Although DIPRO offers an automated fragment recognition for NCI fragments, you might need to specify the fragments by hand sometimes. This is possible via the xcontrol file and the keyword $split. Read in the xcontrol as usual using -I/--input.
$split
fragment1: 1-12,24,25
fragment2: 13-23,26,27
$end
Note
DIPRO works exclusively with two fragments! At any other number of found fragments the calculation will abort.
Example: Benzene - Nitrobenzene
This is a very simple example of a cofacial NCI complex of benzene and nitrobenzene.
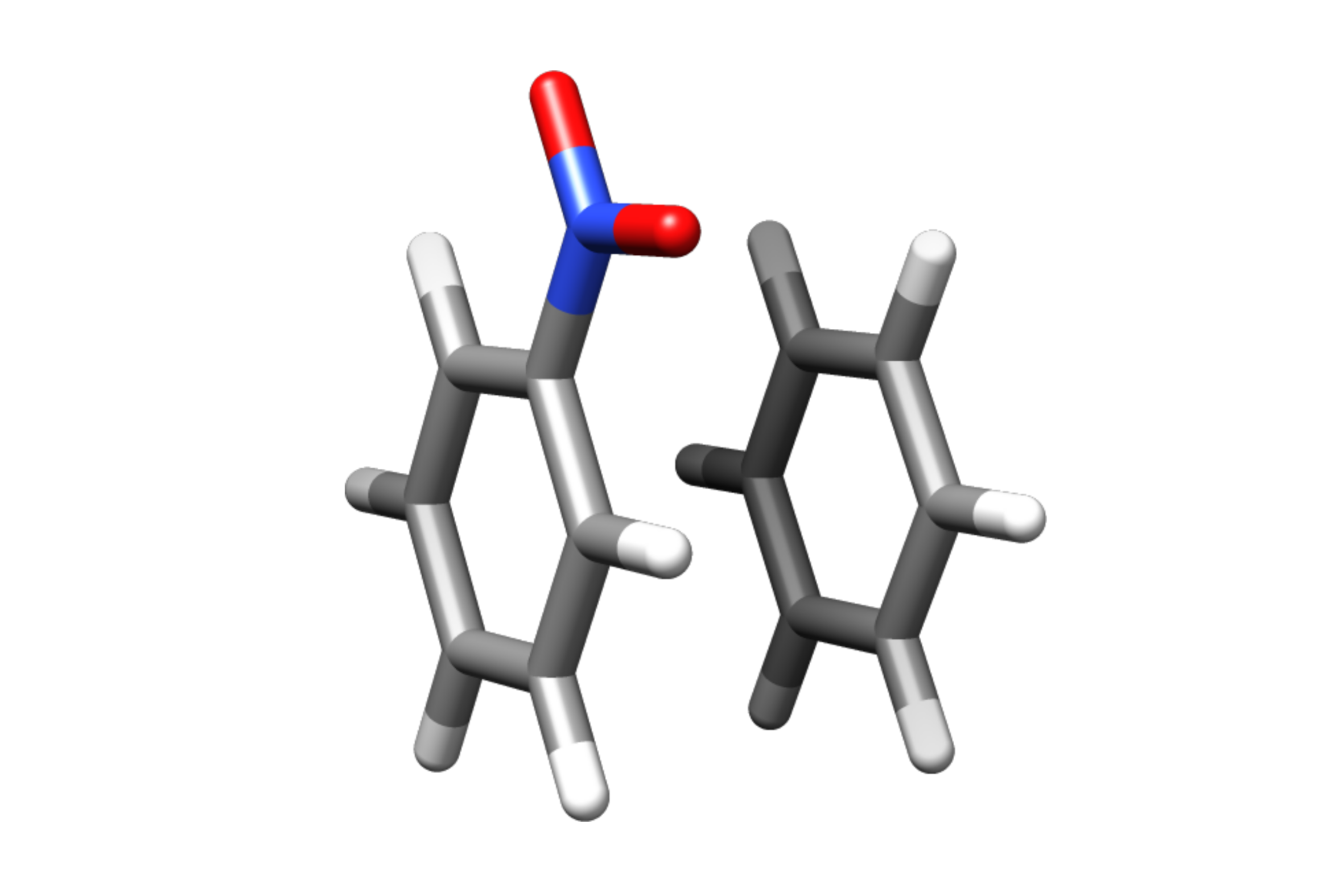
> xtb coord.xyz --dipro --gfn 1
26
C -1.20618 0.69599 1.75211
C -1.20379 -0.70017 1.75215
C 0.00616 -1.39690 1.75210
C 1.21824 -0.70002 1.75171
C 1.23033 0.70989 1.75117
C 0.00125 1.40067 1.75172
H 2.14504 -1.25925 1.74975
H -0.02278 2.48280 1.74976
H -2.14520 1.23405 1.75041
H -2.14028 -1.24255 1.75044
H 0.00554 -2.47919 1.75040
C -1.21072 0.69984 -1.75114
C -1.21074 -0.69902 -1.75149
C 0.00072 -1.39843 -1.75114
C 1.21220 -0.69897 -1.75037
C 1.21222 0.69988 -1.74995
C 0.00077 1.39929 -1.75036
H 2.14941 -1.24008 -1.74839
H 2.14945 1.24099 -1.74755
H 0.00077 2.48148 -1.74837
H -2.14799 1.24091 -1.74971
H -2.14800 -1.24014 -1.75028
H 0.00066 -2.48066 -1.74972
N 2.46723 1.42685 1.74737
O 2.47280 2.62117 1.74468
O 3.50640 0.83800 1.74471
-------------------------------------------------
| D I P R O |
-------------------------------------------------
Calculation for dimer
---------------------
.
.
.
Found 2 fragments!
Fragment 1: 1-11, 24-26
Fragment 2: 12-23
Calculation for fragment 1
------------------------------
charge of fragment : 0
unpaired e- of fragment : 0
.
.
.
Calculation for fragment 2
------------------------------
charge of fragment : 0
unpaired e- of fragment : 0
.
.
.
::::::::::::::::::::::::::::::::::::::::::::::
:: D I P R O C O U P L I N G S ::
::::::::::::::::::::::::::::::::::::::::::::::
energy threshhold for near-degenerate orbitals near HOMO and LUMO considered for DIPRO: 0.100 eV
no. of orbitals within energy window of frag 1 2
no. of orbitals within energy window of frag 2 4
Fragment 1 HOMO Fragment 2 HOMO-1
--------------------------------------
E_mon(orb) frag1 frag2 -12.966 -12.094 eV
|J(AB)|: 0.001 eV
S(AB): 0.00002581
|J(AB,eff)|: 0.000 eV
Fragment 1 HOMO Fragment 2 HOMO
--------------------------------------
E_mon(orb) frag1 frag2 -12.966 -12.099 eV
|J(AB)|: 0.238 eV
S(AB): -0.00998928
|J(AB,eff)|: 0.113 eV
Fragment 1 HOMO Fragment 2 LUMO
--------------------------------------
E_mon(orb) frag1 frag2 -12.966 -7.366 eV
|J(AB)|: 0.002 eV
S(AB): -0.00009192
|J(AB,eff)|: 0.001 eV
Fragment 1 HOMO Fragment 2 LUMO+1
-------------------------------------
E_mon(orb) frag1 frag2 -12.966 -7.361 eV
|J(AB)|: 0.000 eV
S(AB): 0.00001124
|J(AB,eff)|: 0.000 eV
Fragment 1 LUMO Fragment 2 HOMO-1
--------------------------------------
E_mon(orb) frag1 frag2 -9.417 -12.094 eV
|J(AB)|: 0.035 eV
S(AB): -0.00153946
|J(AB,eff)|: 0.019 eV
.
.
.
..............................................................................
: total |J(AB,eff)| for hole transport (occ. MOs) : 0.113 eV :
: total |J(AB,eff)| for charge transport (unocc. MOs) : 0.062 eV :
: total |J(AB,eff)| for charge transfer (CT) : 0.019 eV :
..............................................................................
Please remember, DIPRO is not available for restart!
normal termination of dipro
----------------------------------------------------------
* finished run on 2023/08/25 at 16:43:22.737
----------------------------------------------------------
dipro:
* wall-time: 0 d, 0 h, 0 min, 0.084 sec
* cpu-time: 0 d, 0 h, 0 min, 0.337 sec
* ratio c/w: 3.996 speedup
DIPRO first performs a singlepoint calculation on the complex and yields the fragmentation of your input coordinates. Then, two singlepoint calculations on the fragments structures are performed. The DIPRO output lists which orbitals from the couplings fragments were considered for the coupling calculations, e.g. the HOMO from fragment 1 (here nitrobenzene) and the HOMO-1 from fragment 2 (here benzene), and their corresponding energies. The most important parts are the results for the coupling integrals \(J_{AB}\) and \(J_{AB,eff}\). Furthermore, a total coupling averaged over the square sum of all individual couplings is printed.
Please find more information on the theory and our implementation of DIPRO in our publication (J.Kohn, N.Gildemeister, D.Fazzi, S.Grimme, A.Hansen), Efficient Calculation of Electronic Coupling Integrals with the Dimer Projection Method via a Density Matrix Tight-Binding Potential, J. Chem. Phys. 159, 144106 (2023).