Docking Submodule (aISS)
Note
This feature is only present in version 6.6 and newer or in the current bleeding-edge version.
The automated Interaction Site Screening (aISS) of the docking submodule allows the addition of any molecules to dimers and aggregates.
This is done via an interaction site screening and genetic optimization with the xTB-IFF energy,
followed by GFN geometry optimizations.
By default, only the 15 best structures are optimized, but also an ensemble generation is possible.
The main publication for the aISS can be found at: Angewandte Chemie.
Important
The screening is based on the rigid intermolecular force field xTB-IFF. Thus, changes in the intramolecular geometry are only considered upon geometry optimization.
To run the docking submodule use:
xtb dock [options] <geometry1> <geometry2> [options]
The geometry can be in any format accepted by xtb. If a .CHRG or .UHF file is
present in the current directory, they are read automatically to obtain information about
the charges and number of unpaired electrons of molecules 1 and 2.
They have to contain three lines, starting with the total charge/uhf, followed by
the charge/uhf of molecules 1 and 2.
Options
- --help
Display the help window
- -I, --input <FILE>
Use <FILE> as an input source for
xcontrol(7)instructionsPerforms additionally a pocket search
- --nostack
Switch off the stack search
- --noangular
Switch off the angular search
- --fast
Fast mode for screening and genetic optimization
- --atm
Include ATM term for screening and optimization
- --stepr <REAL>
Radial step size (default = 2.5 Å)
- --stepa <REAL>
Angular step size (default = 45°)
- --maxgen <INT>
Number of cycles for genetic optimization (default = 10)
- --maxparent <INT>
Number of parents for genetic optimization (default = 100)
- --nstack <INT>
Number of gridpoints in one direction of the stack search (default = 1000)
- --nfinal <INT>
Number of final optimizations (default = 15). Setting it to 0 skips the final geometry optimizations.
- --ensemble
Optimizes every structure with a negative xTB-IFF interaction energy and sorts out replicates to yield an NCI ensemble
- --etemp <REAL>
Electronic temperature (default = 300K)
- --iterations <INT>
Maximum iterations for SCF cycle
- -a, --acc <REAL>
Accuracy for SCC calculation, lower is better (default = 1.0)
- --opt <level>
Precision of the optimization. See Geometry Optimization for available options
- --cycles <INT>
Maximum number of optimization cycles
- --gfn2
Perform final optimizations with GFN2-xTB (default)
- --gfn1
Perform final optimizations with GFN1-xTB
- --gfnff
Perform final optimizations with GFN-FF
- --chrg1 <REAL>
Set the total charge of first input structure
- --chrg2 <REAL>
Set the total charge of second input structure
- --uhf1 <REAL>
Set the number of unpaired electrons of first input structure
- --uhf2 <REAL>
Set the number of unpaired electrons of second input structure
- --optlvl <method>
Perform final optimizations with
<method>, e.g., gfn1, gfn2, or gfnff
- –alpb [reference]
analytical linearized Poisson-Boltzmann (ALPB) model, available solvents are acetone, acetonitrile, aniline, benzaldehyde, benzene, ch2cl2, chcl3, cs2, dioxane, dmf, dmso, ether, ethylacetate, furane, hexadecane, hexane, methanol, nitromethane, octanol, woctanol, phenol, toluene, thf, water. The solvent input is not case-sensitive. The Gsolv reference state can be chosen as reference or bar1M (default).
- -g, –gbsa SOLVENT [reference]
generalized born (GB) model with solvent accessible surface (SASA) model, available solvents are acetone, acetonitrile, benzene (only GFN1-xTB), CH2Cl2, CHCl3, CS2, DMF (only GFN2-xTB), DMSO, ether, H2O, methanol, n-hexane (only GFN2-xTB), THF and toluene. The solvent input is not case-sensitive. The Gsolv reference state can be chosen as reference or bar1M (default).
Detailed Input
To read an input file called xtb.inp use
> xtb dock coord1 coord2 --input xtb.inp
In the detailed input, you have control over almost every global variable in the program, some instructions even check your input, but most of the time you should know what you are doing. So in most cases, you can safely rely on the internal defaults or the shipped global configuration file (which should usually be the same). For the final optimizations, many of the standard inputs of xtb will work (Detailed Input) like the constraining and fixing of atoms.
To define variables that belong to the energy screening and genetic
optimization, start a new block in the input file with $dock.
An example might look like this:
$dock
pocket
maxparent = 63
atm
$end
Apart from the general variable, also the directed docking can be activated with an input file. This allows the docking to user-defined regions of molecule 1. The default is an attractive potential for the user-defined atoms, but also a repulsive potential for every other atom can be used (only recommended for large interacting molecules). If the attractive potential is too strong (seen in fusing atoms leading to failing geometry optimizations), a scaling factor can be used to reduce the potential. Values between 0 (no potential) and 1 (default) are recommended. An input file for the directed docking might look like this:
$directed
attractive
scaling factor= 0.9
atoms: 1-5
elements: N
$end
Examples
In the following, two examples are shown of how to perform a docking calculation.
First, we want to start a standard calculation with the following two coordinate inputs
and charges that are placed in a file named .CHRG in the current directory:
26
C 4.91530661517725 6.70283245094063 7.93716475951803
C 4.70274443502525 6.57377729590493 9.29524339877115
H 4.09102174399250 7.26033628697812 9.85619438676986
C 5.30083332347772 5.50886296651214 9.95148435215316
H 5.14950194396918 5.39341270236785 11.01271420108665
C 6.07968625421465 4.60874288641406 9.24518865717228
H 6.54677504050510 3.78278315133684 9.75767540823253
C 6.25703022783366 4.75482454682128 7.88004220926858
H 6.86007029320169 4.04168399010195 7.34301476032045
C 5.66602589617880 5.80800301477451 7.18908033030661
C 5.86757693738733 6.01057612526783 5.69193856008651
C 7.08202831053878 6.91791330345741 5.48228665306979
H 7.24115049352935 7.07340643740184 4.41937035609539
H 7.97015992903950 6.46898272395727 5.91528468405366
H 6.90905459937370 7.88538694516834 5.94891470399975
C 6.00723529207749 4.69862546864148 4.92713759447965
H 5.21841097621933 3.99954166143467 5.19740910959166
H 6.96848369282735 4.23481873803338 5.12168284991214
H 5.95419366234075 4.90262716196177 3.86004767791694
C 3.64840129849507 9.67356063984810 8.63166910176501
O 4.73243266730302 6.72691667725402 5.16545009366973
H 4.06891690953035 6.10561213656021 4.82852948175645
F 4.75466800938595 9.73439880907246 9.35215456436095
F 2.67459899148865 9.19239065754443 9.38100275774183
F 3.32953186458964 10.86751693409879 8.19537197705647
I 4.01066059229276 8.37336628814393 6.88654737084331
15
C 1.69917908436396 3.16419000234708 5.71715609389680
C 2.60797179763240 5.77666501630793 1.55859710223873
N 3.04393410713759 4.87876887895570 4.08766375461315
O 1.71709471089772 5.74460140297995 5.99119818311252
O 0.49329287309353 4.87672637525144 4.06446686790556
O 2.06112121487995 3.28932161619064 2.35293444108821
O 4.39145876797790 4.00283070449141 2.20140862554339
F 2.78451881723356 3.06550089656539 6.49714305953822
F 0.63418103893843 3.02555503292592 6.52249893622828
F 1.70857427523024 2.10779486104579 4.90543392667151
F 1.42635733996611 6.33666447079787 1.81496418774220
F 3.53272631929243 6.73443277485248 1.71239388025687
F 2.60933613238697 5.44729166600378 0.25762027938529
S 1.67585920791859 4.85884119332730 4.86637940756559
S 3.05359431305024 4.27861510795718 2.63234125421372
0
1
-1
The program can then be invoked with:
xtb dock molecule1.xyz molecule2.xyz
It starts with a printout of the calculation setup:
-------------------------------------------------
| Calculation Setup |
-------------------------------------------------
program call : xtb dock molecule1.xyz molecule2.xyz
omp threads : 12
coordinate file A : molecule1.xyz
coordinate file B : molecule2.xyz
number of atoms A : 26
number of atoms B : 15
charge of molecule A : 1.0
charge of molecule B : -1.0
spin of molecule A : 0
spin of molecule B : 0
first test random number : 0.24945994848576
Here, you can check if your molecules, the charge and the spin are read correctly. Next, the computation of electronic properties that are required for the xTB-IFF start:
Precomputation of electronic porperties
For Molecule 1
Successful
System1: Nat:26 Nlmo:45
For Molecule 2
Successful
System2: Nat:15 Nlmo:46
LUMO energy 1 (read) : -11.747
HOMO energy 1 (read) : -15.700
LUMO energy 2 (read) : -1.118
HOMO energy 2 (read) : -8.280
The HOMO and LUMO energies of both molecules are printed. Next, the screening starts:
==============================================
| Starting Energy Screening |
==============================================
Fast Mode selected (recommended)
If ATM term should be included, use -atm option.
Method for final opts. : gfn2
# of genetic optimizations: 10
# of parents : 100
# of final geo. opts. : 15
Rare gas grid step size : 2.50
ang step size /deg : 45.00
# angular grid points : 512
Performing stack search
Performing angular search
initialization done
Total gfn2 energy molecule 1: -48.6147678106
Total gfn2 energy molecule 2: -58.6279172500
A summary of the settings is printed and a single-point calculation for both molecules is performed. The grid-based screening yields a set of starting structures with the best xTB-IFF interaction energies printed:
-----------------------------
Grid based energy screening
-----------------------------
# probe RG points :20384
Best rare gas probe energy/kcal : -2.17
+0.1 charged probe energy/kcal: 0.32
-0.1 charged probe energy/kcal: -10.05
Starting stack search
Grid points: 56000
lowest found /kcal : -179.89
Starting angular search
Grid points:33792
Interaction energy of lowest structures so far in kcal/mol:
-223.79
-179.89
-157.28
-127.74
-119.84
-100.97
-75.50
-73.99
-71.16
-68.25
The best structures are used for the genetic optimization algorithm that runs in multiple cycles. The best and the average xTB-IFF interaction energies are printed for each cycle:
------------------------------
genetic optimization algorithm
------------------------------
cycle Eint/kcal/mol average Eint
1 -307.6 -77.2
2 -347.8 -95.5
3 -364.1 -120.1
4 -364.1 -144.7
5 -385.5 -156.3
6 -385.5 -167.6
7 -385.5 -178.6
8 -395.3 -185.6
9 -395.3 -197.5
10 -395.3 -197.5
Lastly, the structures are optimized and the resulting GFN2-xTB interaction energies are printed:
Optimizing 15 best structures with gfn2
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
---------------------------
Interaction energies
---------------------------
# E_int (kcal/mol)
1 -108.35
2 -105.42
3 -104.13
4 -103.29
5 -97.77
6 -97.08
7 -91.29
8 -87.21
9 -72.18
10 -57.62
11 -55.13
12 -52.83
13 -51.76
14 -49.34
15 -49.34
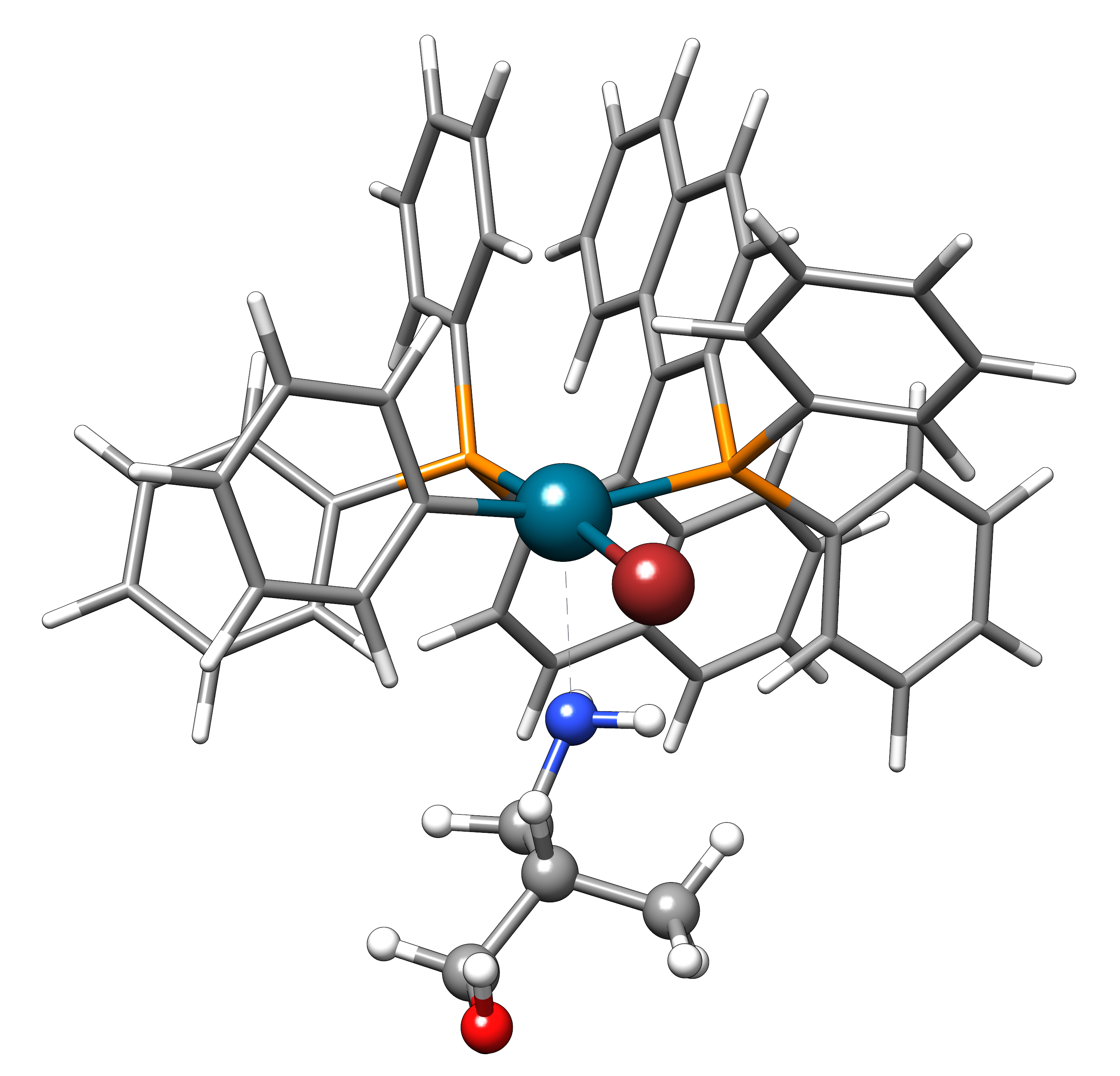
The second example is the use of the directed interaction site screening feature. For this, we have a look at the Buchwald-Hartwig amination and want to add an amine to the catalyst. The normal run-mode with
xtb dock amine.xyz cat.xyz --alpb dmso
17
C -3.83142 2.84076 -0.12858
C -2.71271 3.80734 0.30971
H -3.71462 1.86884 0.40191
H -3.75622 2.64976 -1.22212
N -5.15092 3.38956 0.17246
C -1.33694 3.16769 0.04220
H -1.25577 2.18168 0.55506
O -0.29754 4.00686 0.46991
H -1.20946 2.97517 -1.04499
C -2.83420 5.15491 -0.42082
H -2.80779 3.98502 1.40408
H -3.81369 5.63034 -0.20316
H -2.74149 5.01454 -1.51927
H -2.04024 5.85352 -0.08200
H -0.26205 3.93413 1.45941
H -5.25016 3.49470 1.20852
H -5.87559 2.70758 -0.14876
91
C 1.93043500098766 1.88705038720360 1.27636508509218
C 1.97459955939123 0.74829779266863 0.44541875684329
C 1.02990478561238 -0.25192813715073 0.61271403691281
C 0.01601735664743 -0.12897843953165 1.59684329718372
C 0.00486602136491 1.01110036222777 2.44153841945833
C 0.97835673662409 2.01174361389064 2.24665778060226
C -0.97905754314325 1.11228749020351 3.44591237208907
C -1.91732132645756 0.13258429215518 3.60623449046156
C -1.91580023210662 -0.98853603691249 2.76273320705236
C -0.97388551841309 -1.11865256011331 1.78244381763454
H 2.65978989329656 2.67284509679964 1.13525202046656
P 3.40713569692185 0.45063598133824 -0.66730537531161
H 0.96019342902486 2.88618786841156 2.88182270064755
H -0.97520803758161 1.98254339068831 4.08664095236231
H -2.66738870368542 0.21483103956499 4.37886209692111
H -2.66684386400195 -1.75316672163424 2.89574895976153
H -0.97655866734491 -1.98426023003818 1.13659299566919
C 0.14526976821989 -1.59943917510233 -1.31351060703041
C 1.07251821082195 -1.45287954419223 -0.25056729192353
C 2.01675163349715 -2.43347387046616 -0.00496289247722
P 3.16338763564251 -2.18383389437502 1.40106621266299
C 2.09382972162840 -3.55943079098827 -0.84871958824212
H 2.83641281317833 -4.31953291581165 -0.65217511543419
C 1.24280991573976 -3.69290879737754 -1.90717437340130
H 1.31553262831252 -4.55229356826996 -2.55820916660748
C -0.87091005687256 -0.65166269717626 -1.56151193975196
H -0.95748963139948 0.20553325193108 -0.91031863064544
C -1.73979297136593 -0.81645211711138 -2.60279427063404
H -2.51734053271247 -0.08796162352007 -2.77956466580218
C -1.62876259469206 -1.92603748517661 -3.45412086454612
H -2.31889204202597 -2.03459993938128 -4.27768220744122
C -0.65665910714600 -2.86135403786885 -3.23917435001203
H -0.56554282455158 -3.72146199672846 -3.88685605699420
C 0.24636505357355 -2.72958028321865 -2.16482109262523
H 5.04053258549462 1.95308963427405 1.04874130293099
C 4.89299905597580 2.58285906489136 0.16680216541704
C 4.10488718080303 2.11753771222348 -0.88819970805292
C 3.97295738904971 2.88967645799337 -2.03513257239657
H 3.39723484500199 2.52608137901539 -2.87409316269410
C 4.59842473761614 4.12259113716083 -2.10916621820447
H 4.49632983073021 4.71832348999157 -3.00459307720805
C 5.36052464719349 4.58801603121167 -1.05134179967215
H 5.85161053486878 5.54723300913738 -1.12057939847728
C 5.51132642475435 3.81457877063409 0.08906544598702
H 6.12430221283919 4.16423408293853 0.90645144171876
H 4.02235854811705 -1.50514117966998 -2.62568631546343
C 3.17171560446993 -0.95856763004698 -3.01734431858295
C 2.63287562149439 0.08153169594252 -2.26955458530146
C 1.54867283380819 0.80195882229825 -2.75892383744741
H 1.12910258822322 1.60673248403452 -2.16984850528478
C 1.01257926858250 0.47853247540941 -3.99057124693866
H 0.17235393384064 1.03814430156328 -4.37523265288449
C 1.54560532773241 -0.56581734842844 -4.72987768534842
H 1.11691409607470 -0.81711771999023 -5.68872690644839
C 2.62541313998850 -1.28431754891050 -4.24489077872467
H 3.04105424986400 -2.09553801038726 -4.82408759676300
H 1.33583677784850 -0.82873466427346 5.74959205741078
C 1.26250407321549 -1.53427439323018 4.93563022334869
C 0.29685622096144 -2.52577568384178 4.96085114639427
H -0.38313849657341 -2.59518745232414 5.79689008875041
C 0.19668480503760 -3.42844758612526 3.91363117399716
H -0.55933008558367 -4.19984014517295 3.93572836724455
C 1.05679203332485 -3.33904298939608 2.83622868708840
H 0.97445240569863 -4.03019593470187 2.00770835752968
C 2.03445785679514 -2.34902776756531 2.81406458864911
C 2.13344773754576 -1.44693938519777 3.86489629972164
H 2.89614915297169 -0.67987095722238 3.83313387572892
H 3.25921334801924 -4.71335584827297 3.07226012100153
C 4.00261823026728 -4.76527466002177 2.28923954719084
C 4.13222360890273 -3.72488308629829 1.37980089608449
C 5.12501838704103 -3.78362597909007 0.39615117544011
H 5.25174418682844 -2.94381011451806 -0.29798750601985
C 5.94979789753754 -4.88747721429718 0.30547426182948
H 6.71495850178344 -4.92841143016068 -0.45555496637095
C 5.80628422003005 -5.93092016593784 1.20919590786652
H 6.45742194745829 -6.79014207166281 1.14714340803080
C 4.84174527360330 -5.86441400163025 2.19959273526782
H 4.74234127515449 -6.67004520297639 2.91238124196596
Pd 4.78192197941655 -0.66933638538210 0.85762028033921
Br 6.90486545126967 -0.17867263571098 -0.18326172511155
H 7.37446354631147 -1.04019934049166 6.04329576704061
C 6.91663995455383 -1.04349587877704 5.06457569303456
C 6.05508853955129 -0.02279064401079 4.70077220142163
H 5.83860591747490 0.78008768332450 5.39255355348365
C 5.47218706806705 -0.03062310789953 3.44297757635991
H 4.79334920330727 0.78885028511248 3.17067260451879
C 5.74792721423441 -1.05736492435150 2.54875260346842
C 6.62248653363798 -2.06747078693591 2.90806661030017
H 6.87538232643100 -2.85581054435732 2.20921648721850
C 7.20078991262123 -2.05881341616422 4.16869328286306
H 7.88469613172747 -2.84885038665529 4.44540467384734
will yield a structure with the alcohol moiety bound to the catalyst:
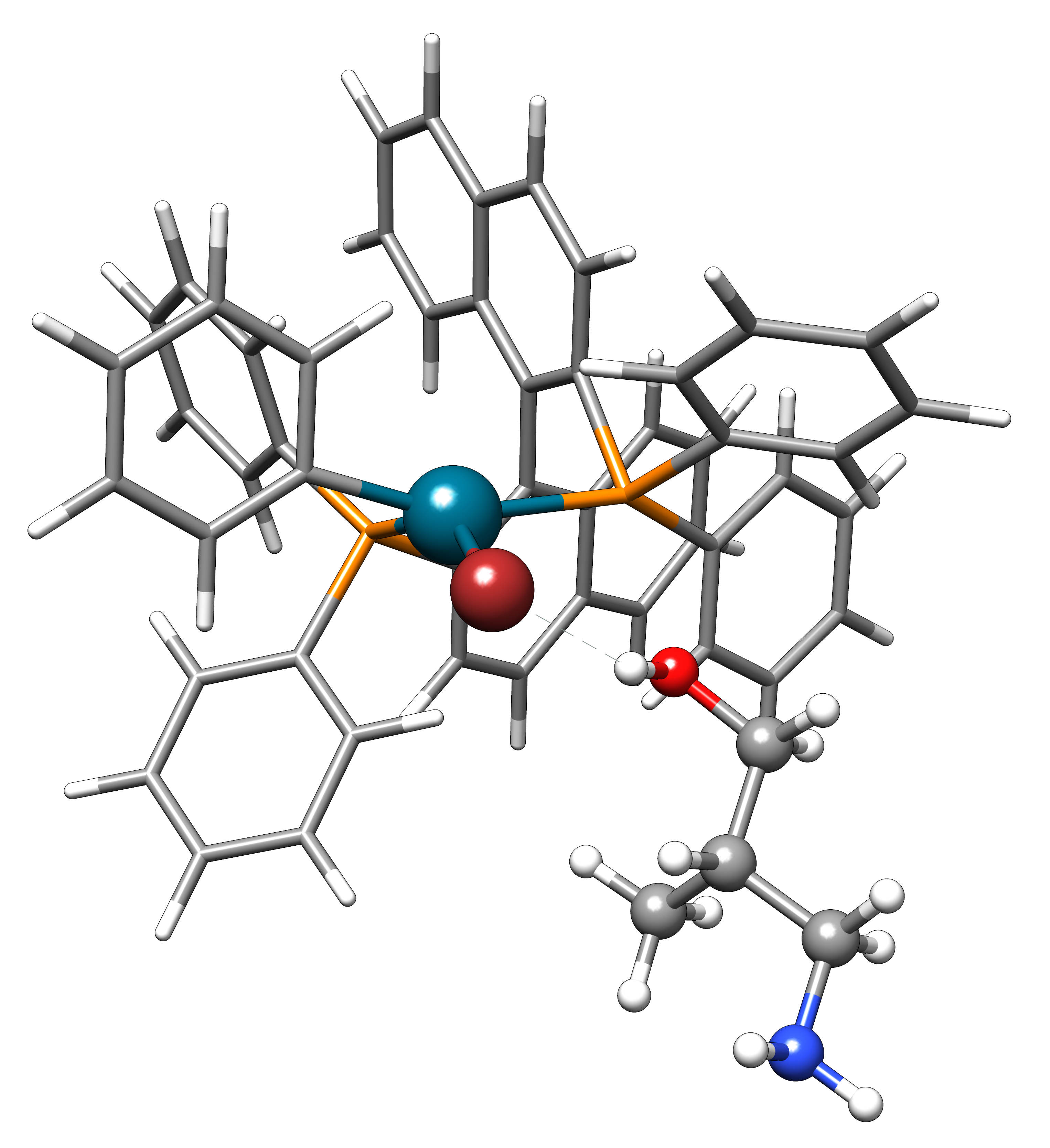
Now, we want to add the amine selectively and thus provide the following xtb.inp input file:
$directed scaling factor= 1.0 atoms: 5,16,17 $end
The scaling factor can be used to adjust the attractive potential in a multiplicatively fashion.
The default is 1.0. A value greater than 1.0 increases the potential, a value lower than 1.0 decreases it.
After invoking xtb with
xtb dock amine.xyz cat.xyz --alpb dmso --input xtb.inp
a structure results where the amine moiety is bound to the catalyst, as proposed for the mechanism: