Usage
cis-4-hexen-1-ol
Example of calculating the 1H-NMR spectrum of cis-4-hexen-1-ol in CHCl3 at 400 MHz.
> cat coord
$coord
-4.5787202885 -0.8553740252 1.3686940377 C
-3.4023148672 -0.1930996279 -1.1104654057 C
-1.3502307141 1.1896725766 -1.5219716436 C
0.3006494795 2.4620980359 0.3955352829 C
0.2362191223 4.5001215909 0.0503890823 H
3.0642161355 1.6025835718 0.2096773622 C
3.4178595291 -1.1774351958 0.9222138508 C
2.5415191057 -2.8708599609 -0.9480135970 O
0.8099880164 -2.4078504756 -1.3346662145 H
5.4277591776 -1.6058314070 1.1410607567 H
2.4658368243 -1.5681688895 2.7281288136 H
4.2123967195 2.7454371194 1.4856132673 H
3.7614131313 1.8836292778 -1.7127605110 H
-0.3830548458 2.1258051759 2.3091187341 H
-0.7518930894 1.4941446048 -3.4579714157 H
-4.3910561682 -0.9675515200 -2.7291675973 H
-3.5318795176 -0.1003881407 2.9630921609 H
-6.5027613245 -0.1227486813 1.4413316443 H
-4.6902514935 -2.9039535570 1.5605291206 H
$end
Input structure.
Start with your (at best already optimized) input structure and create the conformers
and rotamers for the ENSO and ANMR calculation.
> crest coord -gfn2 -g chcl3 -T 4 -nmr > crest.out
In our case CREST found 51 conformers within an energy window of 6 kcal/mol.
We then create a new folder for the ENSO reranking and copy the necessary files:
mkdir enso
cp crest_conformers.xyz coord anmr_nucinfo anmr_rotamer enso/
cd enso/
ENSO requires only the file crest_conformers.xyz, but ANMR needs the last
three files. In the new folder we create the file flags.dat and adapt it to our
choosing. Here we want to calculate everything with TURBOMOLE using PBEh-3c and
DCOSMO-RS for geometry optimization:
enso.py --solvent chcl3 --program tm --programpart4 tm --gfnversion gfn2
--ancoptoptimization on --functional pbeh-3c --solventmodel dcosmors
Tip
You don’t need to give all information using the commandline input and can simply run enso.py without any arguments. This will create the file flags.dat and you can change all information there. But using the commandline input reassures that you don’t have any typos.
> cat flag.dat
FLAGS
nconf: all # all or integer between 0 and total number of conformers
charge: 0 # integer
unpaired: 0 # integer
solvent: chcl3 # acetone, acetonitrile, chcl3, ch2cl2, dmso, h2o, methanol, thf, toluene, gas
prog: tm # tm, orca
ancopt: on # on, off
prog_rrho: xtb # xtb, prog
gfn_version: gfn2 # gfn1, gfn2
temperature: 298.15 # in Kelvin
prog3: tm # tm, orca, prog
prog4: tm # prog, tm, orca
part1: on # on, off
part2: on # on, off
part3: on # on, off
part4: on # on, off
boltzmann: off # on, off
backup: off # on, off
func: pbeh-3c # pbeh-3c, b97-3c, tpss
func3: pw6b95 # pw6b95, wb97x, dsd-blyp
basis3: def2-TZVPP #
couplings: on # on, off
funcJ: pbe0 # tpss, pbe0
basisJ: default #
shieldings: on # on, off
funcS: pbe0 # tpss, pbe0
basisS: default #
part1_threshold: 4.0 # integer or real number
part2_threshold: 2.0 # integer or real number
sm: dcosmors # cosmo, dcosmors, cpcm, smd
smgsolv2: sm # sm, cosmors
sm3: dcosmors # dcosmors, cosmors, smd
sm4: cosmo # cosmo, cpcm, smd
check: on # on, off
crestcheck: off # on, off
maxthreads: 1 # integer larger than 0
omp: 4 # integer larger than 0
reference for 1H: TMS # TMS, DSS
reference for 13C: TMS # TMS, DSS
1H active: on # on, off
13C active: off # on, off
19F active: off # on, off
31P active: off # on, off
resonance frequency: 400 # integer
end
Information on each setting can be found in Settings in .ensorc and flags.dat. Once you have adapted your flags.dat file we can run an input check, testing if the information in flags.dat is correct (no typos and settings combinations are ok {eg. TURBOMOLE doesn’t work with the solvent model SMD}). To do so run:
> enso.py --checkinput
__________________________________________________
| |
| |
| ENSO - |
| energetic sorting of CREST CRE |
| for automated NMR calculations |
| University of Bonn, MCTC |
| July 2018 |
| version 1.26 |
| F. Bohle, K. Schmitz, J. Pisarek and S. Grimme |
| |
|__________________________________________________|
.ensorc is taken from: /home/bohle/.ensorc
Reading absolute paths of programs employed in ENSO.
The following pathways were read in:
ORCA: /tmp1/orca_4_1_0_linux_x86-64_openmpi215
GFN-xTB: /home/abt-grimme/AK-bin/xtb
CREST: /home/bohle/bin/crest
mpshift: /home/bohle/bin/xmpshift
escf: /home/bohle/bin/xescf
Set up of COSMO-RS:
ctd = BP_TZVP_C30_1601.ctd
cdir = "/software/cluster/COSMOthermX16/COSMOtherm/CTDATA-FILES"
ldir = "/software/cluster/COSMOthermX16/COSMOtherm/CTDATA-FILES"
Using /software/cluster/COSMOthermX16/COSMOtherm/DATABASE-COSMO/BP-TZVP-COSMO
as path to the COSMO-RS DATABASE.
Reading user set defaults.
Using conformers from file crest_conformers.xyz.
Using cefine from /home/bohle/bin/cefine
PARNODES for TM or COSMO-RS calculation was set to 4
-----------------------------------------------------------
PARAMETERS
-----------------------------------------------------------
number of atoms in system: 19
number of conformers: 51
charge: 0
unpaired: 0
solvent: chcl3
program for part1 and part2: tm
program for part 3: tm
program for part 4: tm
using ANCOPT implemented in GFN-xTB for the optimization: on
program for RRHO in part 2 and part 3: xtb
temperature: 298.15
GFN-xTB version for RRHO in part 2 and part 3: gfn2
part 1: on
part 2: on
part 3: on
part 4: on
only boltzmann population: off
calculate backup conformers: off
functional for part 1 and 2: pbeh-3c
functional for part 3: pw6b95
basis set for part 3: def2-TZVPP
calculate couplings: on
functional for coupling calculation: pbe0
basis set for coupling calculation: def2-TZVP
calculate shieldings: on
functional for shielding calculation: pbe0
basis set for shielding calculation: def2-TZVP
threshold for part 1: 4.0 kcal/mol
threshold for part 2: 2.0 kcal/mol
solvent model for part 1 and part 2: dcosmors
solvent model for Gsolv contribution of part 2: sm
solvent model for part 3: dcosmors
solvent model for part 4: cosmo
cautious checking for error and failed calculations: on
checking the DFT-ensemble using CREST: off
maxthreads: 1
omp: 4
calculating spectra for: 1H
reference for 1H: TMS
reference for 13C: TMS
reference for 19F: CFCl3
reference for 31P: TMP
resonance frequency: 400
END of parameters
Input check is finished. The ENSO program can be executed with the flag -run.
Please read possible errors at the beginning of the output carefully, they will
hint if your external programs are set up correctly. If you run the ENSO
calculation on a cluster, make sure that all programs are setup correctly in
your jobscript (see Running on a cluster).
Before starting the ENSO calculation, we have to set the number of threads
(maxthreads) (individual calculations e.g. single-points) and number of cores
each thread should have (omp). The total number of cores (maxthreads * omp)
available on your computer is not to be exceeded!
These settings can be written to the global configuration file (if you calculate
always on the same machines) or can be adapted for the individual calculation in
the local configuration file flags.dat.
Tip
When running enso on a cluster it is necessary to source and/or export paths for all employed programs!
Now enso can be run with:
> enso.py -run > enso.out 2> error.enso &
In the following, only parts of the output are shown to highlight important information. During this run in part1 (crude optimization) 49 conformers are within an energy threshold of 4 kcal/mol. These conformers are considered for the full optimization in part2. The two remaining conformers are between 4 and 6 kcal/mol and are considered as backup-confomers. Information of backup conformers is stored in the file enso.json. Since the potential hypersurface between GFNn-xTB and DFT needs not to be parallel, in some cases conformers can be high lying at GFNn-xTB level and low lying at DFT level. To have an automated way of including conformers which would otherwise be disgarded, the ‘backup’ function is introduced and can be activated (after the first enso-run, if it is apparent that dominant conformers are missing) in the local configuration file flags.dat. For this purpose simply restart your enso calculation in the same folder and only change backup: on in the file flags.dat. Then all conformers which are denoted backup-conformers (this information is saved in the file enso.json) will be optimized and evaluated in the sorting procedure.
In part2 first the full DFT optimization is performed and thermostatistical and
solvation contributions are calculated to obtain low-level free energies.
All conformers below a free energy threshold of 2 kcal/mol are considered further.
In this run 35 conformers are considered for part3 and 14 conformers are marked as
backup conformers of part2. After the full optimization at DFT level it is possible
that some conformers could have become rotamers or identical conformers.
This would influence the Boltzmann weights and increase computational cost
unnecessary. To identify them the CREST program is called:
Checking if conformers became rotamers of each other during the DFT-optimization.
The check is performed in the directory conformer_rotamer_check.
Calling CREST to identify rotamers.
WARNING: The following conformers are identified as rotamers or identical.
WARNING: They are NOT sorted out since crestcheck is switched off.
CONFA E(A): G(A):<--> CONFB E(B): G(B):
CONF6 -310.39382 0.43 <--> CONF16 -310.39386 0.34
CONF20 -310.39317 0.50 <--> CONF17 -310.39321 0.96
CONF39 -310.39260 1.08 <--> CONF38 -310.39269 0.96
CONF46 -310.39195 1.70 <--> CONF13 -310.39196 2.03
Since the crestcheck flag is switched off, the conformers are not automatically sorted out and the user has to visually inspect the molecules and sort the conformers as described in Sorting out rotamers at DFT level detected by CREST. We advise you to sort out conformers manually.
In part3 high level DFT single-points are calculated on the 35 conformers passed
from part2. The boltzmann weights are calculated and 24 conformers are considered
for the NMR property calculations in part4. The ‘file populated-conf-part3.xyz’ is written at
the end of part3 and contains all populated conformers. The file ‘anmr_enso’ is written
which is read from ANMR and provides the Boltzmann weights of the populated conformers.
In part4 the couplings and shielding calculations were performed. Finally the file .anmrrc file is written.
At the very end ENSO checks if possible rotamers of each other are in the populated ensemble and notifies the user:
writing .anmrrc
Results are written to enso.json.
***---------------------------------------------------------***
Printing most relevant errors again, just for user convenience:
Possible rotamers of each other still in final ensemble: CONF39 <--> CONF38. Please check by hand!
Possible rotamers of each other still in final ensemble: CONF20 <--> CONF17. Please check by hand!
Possible rotamers of each other still in final ensemble: CONF6 <--> CONF16. Please check by hand!
***---------------------------------------------------------***
END of part4.
-----------------------------------------------------------
ENSO all done!
After inspecting the conformers CONF39 is removed from the ensemble (details on how to do that are shown in Sorting out rotamers at DFT level detected by CREST), because I considered it to be identical to CONF38. After removing the identical conformer we can restart enso.
> enso.py -run > enso.out 2> error.enso &
This time enso will read all necessary information from the file enso.json and
operate with this data. At the end of part3 the Boltzmann weight is recalculated
and two conformers are now additionally populated. For them coupling and shielding
constants are calculated in part4. All files for ANMR are updated.
Now all information is present and ANMR can be called to calculate the full
NMR spectrum:
> anmr > anmr.out 2> error.anmr &
+--------------------------------------+
| A N M R |
| S. Grimme |
| Universitaet Bonn, MCTC |
| 1989-2019 |
| version 3.5 |
| Sat Feb 9 06:41:57 CET 2019 |
+--------------------------------------+
Based on a TurboPascal program written
in 1989 which was translated to F90 in
2005 and re-activated in 2017.
Please cite work employing this code as:
ANMR Ver. 3.5: An automatic, QC based
coupled NMR spectra simulation program.
S. Grimme, Universitaet Bonn, 2019
S. Grimme, C. Bannwarth, S. Dohm, A. Hansen
J. Pisarek, P. Pracht, J. Seibert, F. Neese
Angew. Chem. Int. Ed. 2017, 56, 14763-14769.
DOI:10.1002/anie.201708266
=============================
# OMP threads = 1
=============================
reading <.anmrrc> for standard data
Read TM from anmrrc
1 31.7860000000000 0.000000000000000E+000 1
6 189.674000000000 0.000000000000000E+000 0
9 182.570000000000 0.000000000000000E+000 0
15 291.900000000000 0.000000000000000E+000 0
1H resonance frequency (-mf <real>) : 400.00
line width (-lw <real>) : 1.00
number of all conformers :25
remove J couplings to OH groups : T
maximum spin system size in a fragment :14
fragmentation type (0=none,1=at,2=mol) : 2
chemical shift scalings a,b : 1.00 0.00
spin-spin coupling scal factor : 1.07
plot offset : 0.00
Active nuclei :H
...
SKIPPING A LOT OF OUTPUT
...
=== FRAGMENTED SYSTEM ===
====================================================
solving (J/sigma) averaged spin Hamiltonian
====================================================
spinsystem 1 with 9 spins
1024 product functions 12 Mt blocks, largest is 210
1( 1) 2( 9) 3( 37) 4( 93) 5( 162) 6( 210) 7( 210) 8( 162) 9( 93) 10( 37) 11( 9) 12( 1)
first maxtrix multiply, sparsity in % 99.536 ...
second maxtrix multiply, sparsity in % 94.781 ...
512 product functions 10 Mt blocks, largest is 126
1( 1) 2( 9) 3( 36) 4( 84) 5( 126) 6( 126) 7( 84) 8( 36) 9( 9) 10( 1)
first maxtrix multiply, sparsity in % 99.121 ...
second maxtrix multiply, sparsity in % 91.919 ...
done.
12436 non-zero transitions.
spectrum on <anmr.dat>
Range (delta in ppm) 0.437872924804687 7.03675048828125
Range (delta in Hz) 175.149169921875 2814.70019531250
Min/max Int. ) 4.171584807901128E-003
computing spectrum ...
done.
writing output file ...
done.
All done.
After ANMR finished computing, the file anmr.dat is written and it contains
the spectrum (intensity vs shift) the user can plot:
> nmrplot.py -i anmr.dat exp.dat -start 0 -end 6.5 -o 1Hspectrum -orientation 1 -1
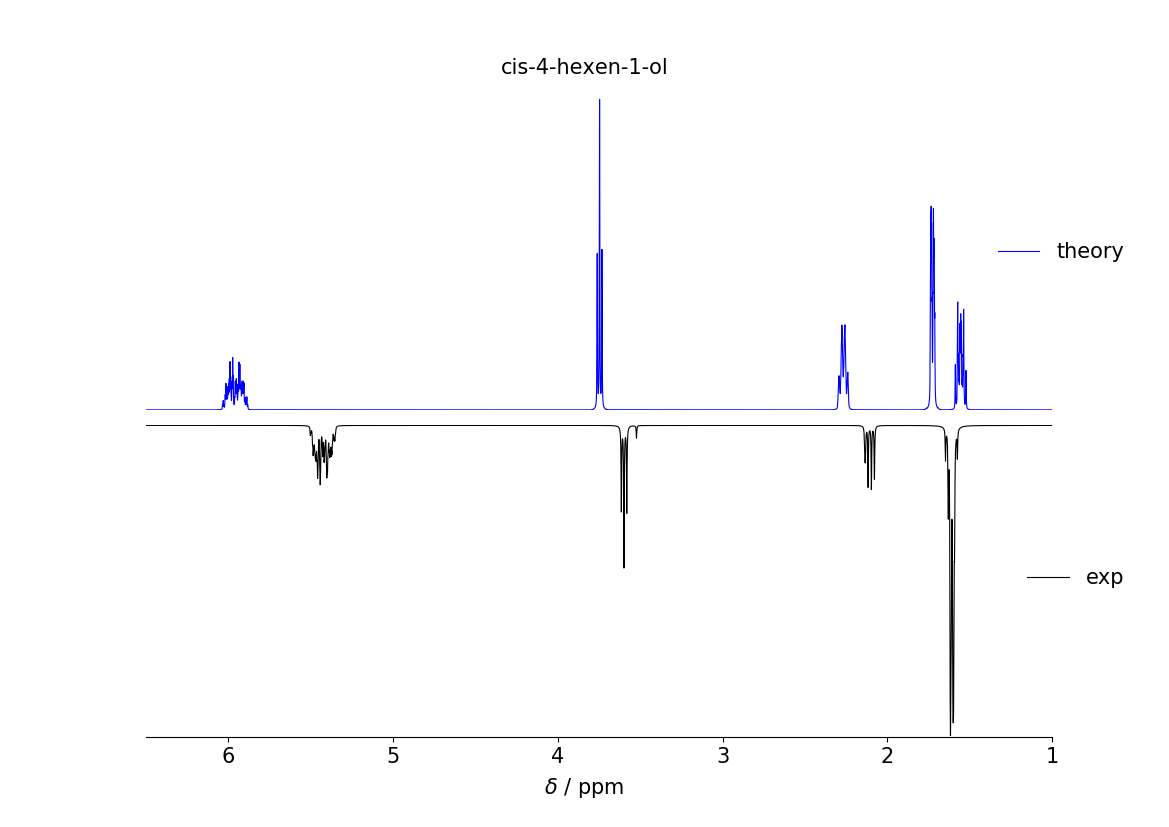
1H NMR spectrum of cis-4-hexen-1-ol in chloroform at 400 MHz, comparing calculated and experimental spectrum. Exp taken from [SDBSWeb : https://sdbs.db.aist.go.jp (National Institute of Advanced Industrial Science and Technology,16-10-2019) (SDBSNo. 11748)].
2-methyl-1-pentene
Example of calculating the 1H-NMR spectrum of 2-methyl-1-pentene in CHCl3 at 400 MHz.
> cat coord
$coord
-5.1134989926 0.0445408597 0.0007215195 C
-2.3988260553 0.1202192416 0.9598504570 C
-2.0426150350 1.9467776447 1.8509773297 H
-0.4955528936 -0.3025973506 -1.1852527430 C
2.1853738985 -0.2583887206 -0.2367582425 C
3.4286190716 -2.3618737092 0.3005853656 C
2.5901373582 -4.2004809628 0.0485727882 H
5.3374488734 -2.3390838060 1.0097369787 H
3.3398174602 2.3079102171 0.0825121447 C
5.2234930962 2.1788391495 0.8913733279 H
2.1708137054 3.4751040066 1.3098219746 H
3.4682822356 3.2543034689 -1.7427970988 H
-0.7536049204 1.1708293724 -2.6081830586 H
-0.8901990516 -2.1258718566 -2.0673390015 H
-2.1284937554 -1.3401645088 2.3937954454 H
-6.4334377217 0.3509962700 1.5473865797 H
-5.4204111085 1.5054637513 -1.4143993805 H
-5.5276306722 -1.7796998127 -0.8540259276 H
$end
Input structure.
Start with your (at best already optimized) input structure and create the conformers
and rotamers for the ENSO and ANMR calculation.
> crest coord -gfn2 -g chcl3 -T 4 -nmr > crest.out
In our case CREST found 6 conformers within an energy window of 6 kcal/mol.
We then create a new folder for the ENSO reranking and copy the necessary files:
mkdir enso
cp crest_conformers.xyz coord anmr_nucinfo anmr_rotamer enso/
cd enso/
ENSO requires only the file crest_conformers.xyz, but ANMR needs the last
three files. In the new folder we create the file flags.dat and adapt it to our
choosing. Here we want to calculate everything with ORCA using PBEh-3c and SMD for
geometry optimization:
enso.py --solvent chcl3 --program orca --programpart4 orca --gfnversion gfn2
--ancoptoptimization on --functional pbeh-3c --solventmodel smd
> cat flag.dat
FLAGS
nconf: all # all or integer between 0 and total number of conformers
charge: 0 # integer
unpaired: 0 # integer
solvent: chcl3 # acetone, acetonitrile, chcl3, ch2cl2, dmso, h2o, methanol, thf, toluene, gas
prog: orca # tm, orca
ancopt: on # on, off
prog_rrho: xtb # xtb, prog
gfn_version: gfn2 # gfn1, gfn2
temperature: 298.15 # in Kelvin
prog3: prog # tm, orca, prog
prog4: prog # prog, tm, orca
part1: on # on, off
part2: on # on, off
part3: on # on, off
part4: on # on, off
boltzmann: off # on, off
backup: off # on, off
func: pbeh-3c # pbeh-3c, b97-3c, tpss
func3: pw6b95 # pw6b95, wb97x, dsd-blyp
basis3: def2-TZVPP #
couplings: on # on, off
funcJ: pbe0 # tpss, pbe0
basisJ: default #
shieldings: on # on, off
funcS: pbe0 # tpss, pbe0
basisS: default #
part1_threshold: 4.0 # integer or real number
part2_threshold: 2.0 # integer or real number
sm: smd # cosmo, dcosmors, cpcm, smd
smgsolv2: sm # sm, cosmors
sm3: smd # dcosmors, cosmors, smd
sm4: cpcm # cosmo, cpcm, smd
check: on # on, off
crestcheck: off # on, off
maxthreads: 3 # integer larger than 0
omp: 4 # integer larger than 0
reference for 1H: TMS # TMS, DSS
reference for 13C: TMS # TMS, DSS
1H active: on # on, off
13C active: off # on, off
19F active: off # on, off
31P active: off # on, off
resonance frequency: 400 # integer
end
After ANMR finished computing, the file anmr.dat is written and it contains
the spectrum (intensity vs shift) the user can plot:
> nmrplot.py -i anmr.dat exp.dat -start 0 -end 6 -o 1Hspectrum -orientation 1 -1
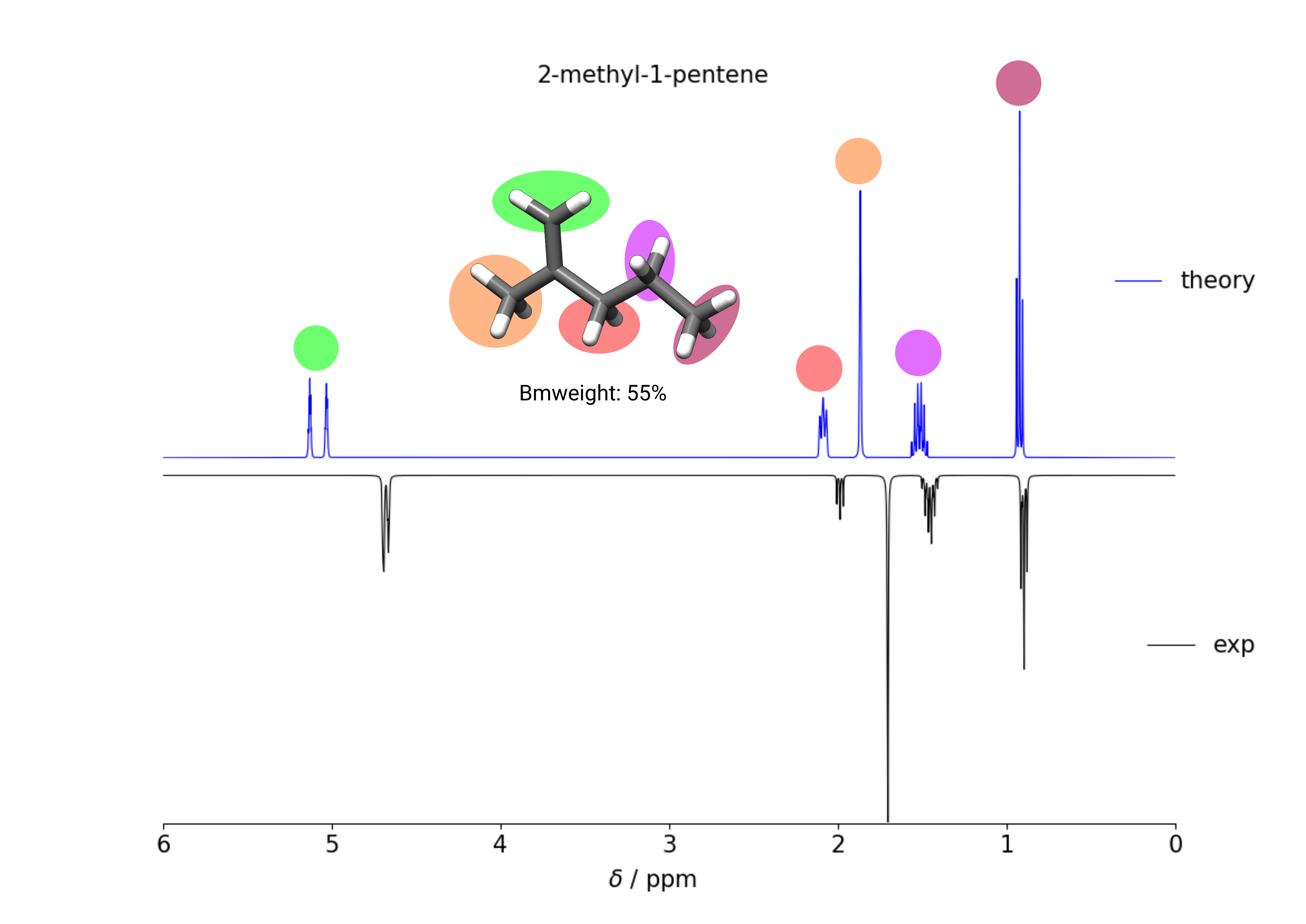
1H NMR spectrum of 2-methly-1-pentene in chloroform at 400 MHz, comparing calculated and experimental spectrum. Exp taken from [SDBSWeb : https://sdbs.db.aist.go.jp (National Institute of Advanced Industrial Science and Technology,16-10-2019) (SDBSNo. 225)].
Manually restarting calculations and modifying enso.json
In the event that an optimization failes, ENSO discardes the geometry and will not
try to reoptimize the structure. The user can optimize the geometry manually and add the
information on convergence (success) and energy (at the same level as all other calculations)
to the file enso.json. When restarting ENSO the information on the conformers of the previous
run is read from the file enso.json and the manually corrected conformer is further evaluated
within the ongoing enso-run.
Here we show an example on how to manually restart an optimization (part1):
'In case of ORCA using the ANCOPT optimizer implemented in xtb:'
write the geometry in form of a coord file and add at the end of the file:
$external
orca input file = inp
$end
The file 'inp' contains the input for the ORCA program eg.:
%MaxCore 8000
! def2-mSVP pbeh-3c grid4
!ENGRAD
! smallprint printgap noloewdin
%output
print[P_BondOrder_M] 1
print[P_Mayer] 1
print[P_basis] 2
end
%pal
nprocs 7
end
%cpcm
smd true
smdsolvent "chloroform"
end
* xyzfile 0 1 inp.xyz
Then convert the tmole coord file into an xmol file (required by the ORCA input):
> t2x coord > inp.xyz
and then run xtb as driver for ORCA:
> xtb coord --opt crude --orca > opt-part1.out
after convergence convert the optimized geometry back to the coord file!
If the optimization is converged the information has to be written to the enso.json file (to be on the save side, first make a copy of your enso.json file). enso.json contains data on each conformer in the following form:
"plain_uncalculated_CONF":
{
"crude_opt": "not_calculated",
"energy_crude_opt": null,
"backup_for_part2": false,
"consider_for_part2": true,
"opt": "not_calculated",
"energy_opt": null,
"backup_for_part3": false,
"sp_part2": "not_calculated",
"energy_sp_part2": null,
"consider_for_part3": true,
"sp_part3": "calculated",
"energy_sp_part3": null,
"cosmo-rs": "not_calculated",
"energy_cosmo-rs": null,
"gbsa_gsolv": "not_calculated",
"energy_gbsa_gsolv": null,
"rrho": "calculated",
"energy_rrho": null,
"symmetry": "c1",
"consider_for_part4": true,
"1H_J": "not_calculated",
"1H_S": "not_calculated",
"13C_J": "not_calculated",
"13C_S": "not_calculated",
"19F_J": "not_calculated",
"19F_S": "not_calculated",
"31P_J": "not_calculated",
"31P_S": "not_calculated"
"removed_by_user": false
},
"CONF1-example":
{
"crude_opt": "calculated",
"energy_crude_opt": -1034.78500028827,
"backup_for_part2": false,
"consider_for_part2": true,
"opt": "calculated",
"energy_opt": -1034.786028019303,
"backup_for_part3": true,
"sp_part2": "calculated",
"energy_sp_part2": -1034.76193327809,
"consider_for_part3": false,
"sp_part3": "not_calculated",
"energy_sp_part3": null,
"cosmo-rs": "calculated",
"energy_cosmo-rs": -0.028947468366750467,
"gbsa_gsolv": "not_calculated",
"energy_gbsa_gsolv": null,
"rrho": "calculated",
"energy_rrho": 0.369622527533,
"symmetry": "c1",
"consider_for_part4": false,
"1H_J": "not_calculated",
"1H_S": "not_calculated",
"13C_J": "not_calculated",
"13C_S": "not_calculated",
"19F_J": "not_calculated",
"19F_S": "not_calculated",
"31P_J": "not_calculated",
"31P_S": "not_calculated",
"removed_by_user": false
},
Now to change the information on the converged crude optimization (for the conformer CONFX), you have to change:
crude_opt: calculated
energy_crude_opt: eg: -1034.780801227157
Using the updated information, restart your enso-run and the program will automatically evaluate how to proceed with the new information(e.g. sort out this conformer or calculate a full optimization…). You can restart your ENSO-run with:
> enso.py -run > enso.out 2> error.enso &
Note
This procedure is not limited to optimizations, but every failed
calculation can be manually performed. Then the result has to be written to
the file enso.json in the same manner. Then ENSO can restart on the
introduced information.
Sorting out rotamers at DFT level detected by CREST
In part2 the conformers are fully optimized at DFT level. It is possible that
during optimization conformers can become rotamers or identical. To identify
these the CREST program is called. If rotamers are found, then ENSO can
sort them out if the crestcheck flag is set. Or the user is simply informed on
the possible existence of these conformers and has to visually inspect the geometries,
decide and adjust the necessary information in the file enso.json (which we recommend).
Here we show which entries to change in the file enso.json:
Note
CREST evaluates conformers to be rotamers or identical based on thresholds concerning the electronic energy, rotational constant and RMSD on the geometries. Visual inspection of possible conformers is encouraged for molecules which show very similar conformers.
To sort out conformers, change the flag consider_for_part3 to false:
"CONFX":
{...
"consider_for_part2": false,
"consider_for_part3": false,
"consider_for_part4": false,
...}
Then set part 1 off within the file 'flags.dat'!
Otherwise consider_for_part_2 will be reset to true ...
Or you can simply set:
"CONFX":
{...
"removed_by_user": true,
...}
This will sort out the conformer.
Running on a cluster
When submitting an enso calculation with a jobscript on a computer cluster it is necessary to source and export all programs or paths needed for the enso calculation! An example jobscript looks like:
#!/bin/bash
# PBS Job
#PBS -V
#PBS -N ENSO-Calculation
#PBS -m ae
#PBS -q batch
#PBS -l nodes=1:ppn=14
#
cd $PBS_O_WORKDIR
### setup programs
## XTB
export OMP_NUM_THREADS=1
export MKL_NUM_THREADS=1
ulimit -s unlimited
export OMP_STACKSIZE=1000m
export XTBHOME=/home/$USER/.XTBPARAM/
## TM
export PARA_ARCH=SMP
source /home/path/TURBOMOLE.7.2.1/turbo721
export PARNODES=4
export TM_PAR_FORK=1
## ORCA4.2.1
ORCAPATH="/tmp1/orca_4_2_1_linux_x86-64_openmpi216";
MPIPATH="/software/openmpi-2.1.5/bin";
MPILIB="/software/openmpi-2.1.5/lib64";
PATH=${ORCAPATH}:${MPIPATH}:$PATH
export LD_LIBRARY_PATH=${ORCAPATH}:${MPILIB}:$LD_LIBRARY_PATH
# PATH
PATH=/home/$USER/bin:$PATH
export PATH
### end programs + PATH
export HOSTS_FILE=$PBS_NODEFILE
cat $HOSTS_FILE>hosts_file
TMP_DIR=/tmp1/$USER
DIR1=$PWD
mkdir -p $TMP_DIR/$PBS_JOBID
rsync -q --bwlimit=5000 $DIR1/* $TMP_DIR/$PBS_JOBID/
rsync -q --bwlimit=5000 $DIR1/.* $TMP_DIR/$PBS_JOBID/
rsync -rq --ignore-missing-args --bwlimit=5000 $DIR1/CONF* $TMP_DIR/$PBS_JOBID/
rsync -rq --ignore-missing-args --bwlimit=5000 $DIR1/NMR* $TMP_DIR/$PBS_JOBID/
cd $TMP_DIR/$PBS_JOBID
####################################################################################
#jobs start here (if you have no idea what this script does, only edit this part...)
export PYTHONUNBUFFERED=1
./enso.py -run > enso.out 2> error.enso
#end of job (....and stop editing here.)
#####################################################################################
#copy everything back that is smaller than 5 gbytes
rsync -rq --bwlimit=5000 --max-size=5G $TMP_DIR/$PBS_JOBID/* $DIR1/
rsync -q --bwlimit=5000 --max-size=5G $TMP_DIR/$PBS_JOBID/.* $DIR1/
rsync -rq --bwlimit=5000 --include="*/" --include="mos" --include="alpha" --include="beta" --exclude=* $TMP_DIR/$PBS_JOBID/* $DIR1/
cd $DIR1
rm -r $TMP_DIR/$PBS_JOBID
Tipps
Using the def2-QZVP(-gf) basis (only possible with TURBOMOLE and the latest cefine version)! When calculating the high level single-point in part3 the basis def2-QZVP(-gf) basis set can be used. Here all f functions are deleted from hydrogen and all g functions from everything else. This is only supposed to speed up the calculation.
Optimization and sorting the CRE only based on the energy (and not free energy). It is possible to not calculate thermostatistical contributions to free energy, by simply setting the programm for prog_rrho to off. This way all calculations for \(G_{RRHO}\) will be skipped.
Troubleshooting
Handling of missing conformers:
Use the backup functionallity and make sure that you are not missing a conformer which is high lying in energy at GFNn-xTB level. (See backup)
The conformer might be wrongly sorted into a different conformer group by CREST and counted as a rotamer. To check this you can resort the complete CRE (including all rotamers and conformers) and check if the thresholds employed in the CREGEN sorting routine changes the CRE.
crest coord -cregen crest_rotamers_6.xyz -bthr 12 -nmr > cregen.out
Here you have to inspect the printout and check if grouping/sorting of conformers is correct. If you find that there were conformers wrongly grouped because of sorting-thresholds not suitable for the investigated system. You have to rerun the ENSO calculation on the new ensemble file.
Unparallel hypersurfaces between the functional for optimization and high level free energy
It is possible that the sorting between the functional/basis used for geometry optimization and the method combination (functional/basis set) for the high level free energy is not parallel. One option is to use a larger basis set for the high level free energy calculation. The default basis set for the single-point calculation in part3 is the def2-TZVP basis. It can be necessary to use a larger basis set. To to so, after the previous run, simply change the basis3 to the basis you desire in the file flags.dat and restart your enso calculation. Be advised that doing so results in the recalculation of all previously calculated single-points in part3!
Influence of the Boltzmann population on the final spectrum
The influence of the Boltzmann population on the final spectrum can be checked either by manually adjusting the populations in the file .anmrrc
or by employing different functionals/basis sets
or by checking different solvation models. A quick test, when comparing to additive solvation models (like COSMO-RS) is to use the flag gbsa_gsolv which will calculate a solvation contribution to free energy and performs rather fast.
by verifying that no conformers have become rotamers or identical to each other during DFT optimization (ENSO will inform you on possible cases, but the evaluation should be performed manually)